R包是多个函数的集合
安装和加载R包
镜像设置
将 CRAN 镜像地址设置为了清华大学的镜像站点。这样,在使用 R 语言安装或更新包时,系统会优先从清华大学的镜像站点下载,从而加快下载速度
options("repos"=c(CRAN="http://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror="http://mirrors.tuna.tsinghua.edu.cn/bioconductor/")
安装
R包安装命令是install.packages()或者BiocManager::install()。具体使用哪一个是取决于你要安装的包存在于CRAN网站还是Biocductor,怎么知道存在于哪里呢?可以谷歌必应搜到的
加载
library和require,两个函数均可。使用一个包,是需要先安装再加载,才能使用包里的函数
dplyr包中函数使用
mutate()
mutate(test, new = Sepal.Length * Sepal.Width)select(),列筛选
#列号
select(test,1)
select(test,c(1,5))
#列名
select(test,Petal.Length, Petal.Width)
vars=c("Petal.Length", "Petal.Width")
select(test,one_of(vars))
filter(),行筛选
filter(test, Species == "setosa"&Sepal.Length > 5
filter(test, Species %in% c("setosa","versicolor"))arrange(),排序
arrange(test, Sepal.Length)#默认从小到大排序
arrange(test, desc(Sepal.Length))#用desc从大到小summarise(),汇总
summarise(test, mean(Sepal.Length), sd(Sepal.Length))先按照Species分组,计算每组Sepal.Length的平均值和标准差
group_by(test, Species)
summarise(group_by(test, Species),mean(Sepal.Length), sd(Sepal.Length))
dplyr包小技巧及数据处理
管道符:cmd/ctr + shift + M
test %>%
group_by(Species) %>%
summarise(mean(Sepal.Length), sd(Sepal.Length))count统计unique
count(test,Species)连接两个表
test1 <- data.frame(x = c('b','e','f','x'),
z = c("A","B","C",'D'))
test2 <- data.frame(x = c('a','b','c','d','e','f'),
y = c(1,2,3,4,5,6))
#取交集
inner_join(test1, test2, by = "x")
#左连接,或者连接其中一个
left_join(test1, test2, by = 'x')
left_join(test2, test1, by = 'x')
#全连接
full_join( test1, test2, by = 'x')
表格匹配
#半连接:返回能够与y表匹配的x表所有记录
semi_join(x = test1, y = test2, by = 'x')
#反连接:返回无法与y表匹配的x表的所记录
anti_join(x = test2, y = test1, by = 'x')表格合并
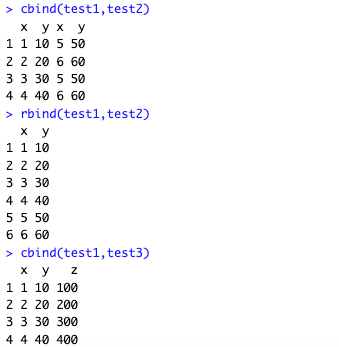
bind_rows(test1, test2)
bind_cols(test1, test3)测试
R中自带的cbind函数和rbind函数