一、红外光谱产生的原理
在不破坏键合的条件下,分子内核-核之间的构型会发生变化,构成分子振动的基础。在偏离核间距不大的情况下,近似为抛物线,对应的振动为简谐振动。下图是两种典型的势能曲线:
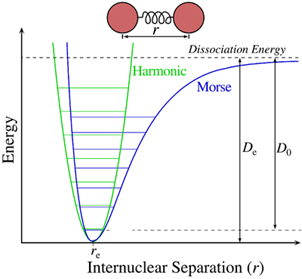
绿线为谐振势,蓝线为更接近实际情况的Morse势。
对简谐振动进行量子力学处理,可得能量为
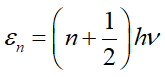
其中n为振动量子数,hν为给定的振动模式的两个相邻能级的能量差。室温下分子一般处于振动基态,吸收红外光子后会发生振动激发,产生红外光谱。
对某振动模式,从基态跃迁到第一激发态吸收的光子的频率称为该振动模式的基频。对于n个原子组成的分子来说,非线形分子有3n−6个振动自由度,线形分子有3n−5个振动自由度。原则上讲,每一个振动自由度都会对应一个吸收峰,但实际上红外吸收峰常少于振动自由度,因为:(1)根据光谱选律,伴随偶极变化的振动才有红外吸收;(2)振动频率相同的振动形式发生简并;(3)仪器分辨率不高,不能将频率接近的吸收峰分开,弱的吸收也有可能检测不出。例如CO2分子,共有4个振动模式,高斯计算后的结果如下(下文会说明如何计算):
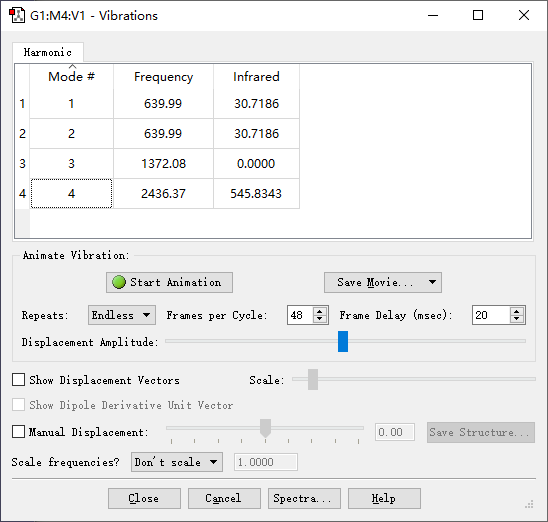
可以看到前两个振动模式是简并的,只有一个吸收,而第三个振动的红外强度为0,也没有吸收,因为它对应着两个C=O的对称伸缩振动,分子的偶极一直保持为0:
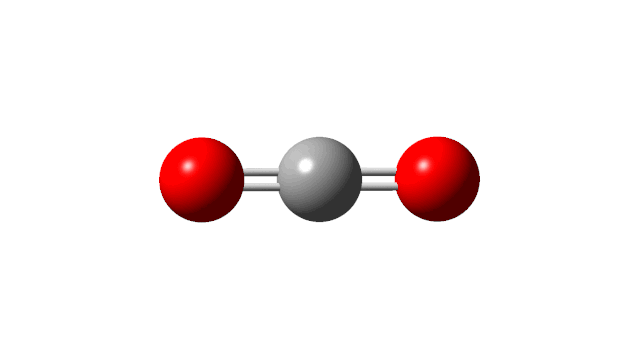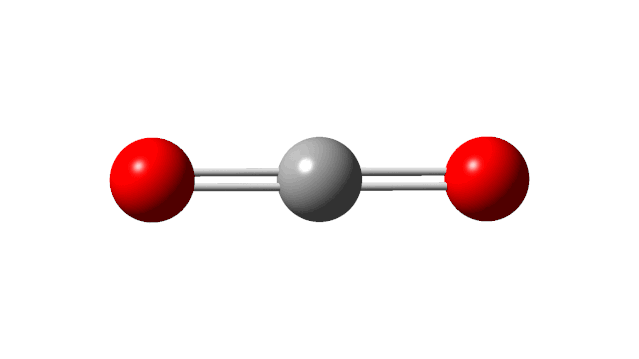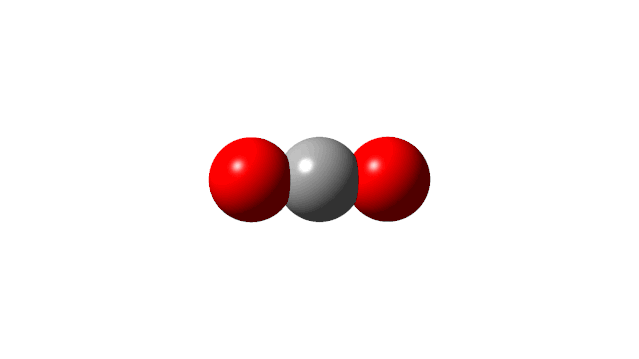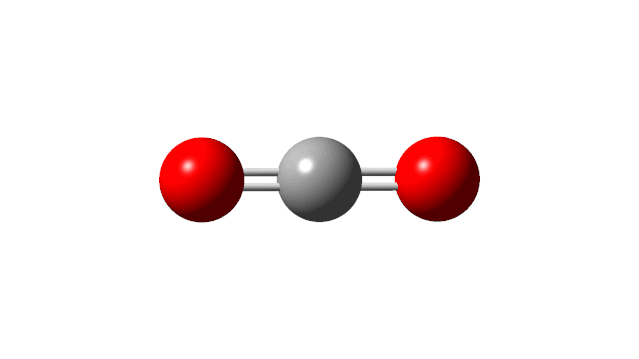
因此,CO2分子共有两个基频。
红外光谱中,振动从基态到第二、第三激发态等产生的吸收峰几乎是基频的整数倍,称为倍频;如果分子吸收一个光子而同时激发两种或两种以上的基频跃迁,称为合频;某些原先被激发的模式会在新的激发过程中将能量传递给新模式,自身返回振动基态,称为差频。合频峰和差频峰统称泛频峰。由于倍频和泛频的存在,有时红外吸收峰会多于振动自由度。
二、红外光谱的计算
计算红外光谱只需要对分子进行频率计算即可,高斯中一般直接使用opt freq的组合。以下以甲醛分子为例,输入文件为
%chk= HCHO.chk #p b3lyp/def2svp opt freqHCHO
0 1
C -1.17400418 -0.52935010 0.00000000
O 0.05331282 -0.52935010 0.00000000
H -1.76614918 0.41005390 0.00000000
H -1.76614918 -1.46875410 0.00003900
用GaussView打开输出文件,选择Results→Vibrations,就可以看到振动频率相关的信息:
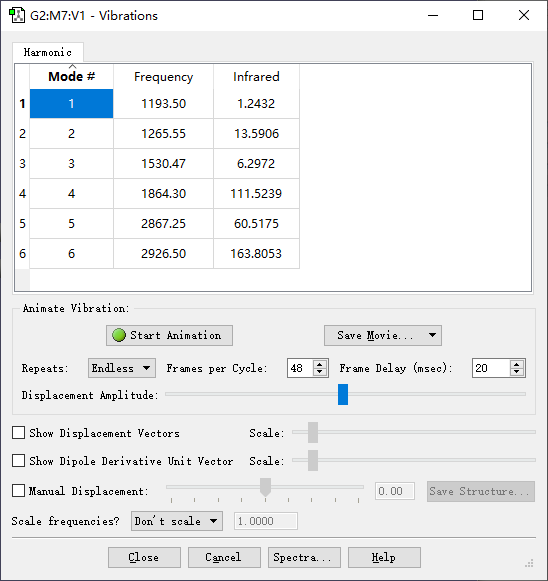
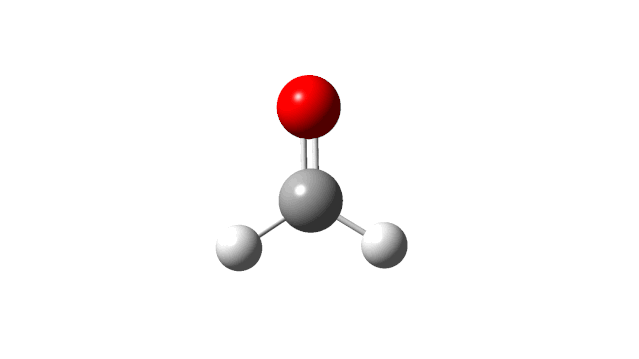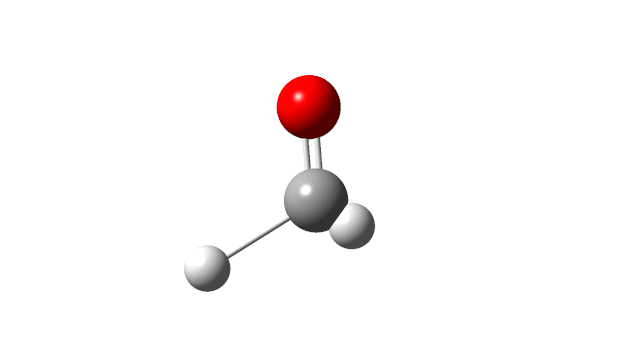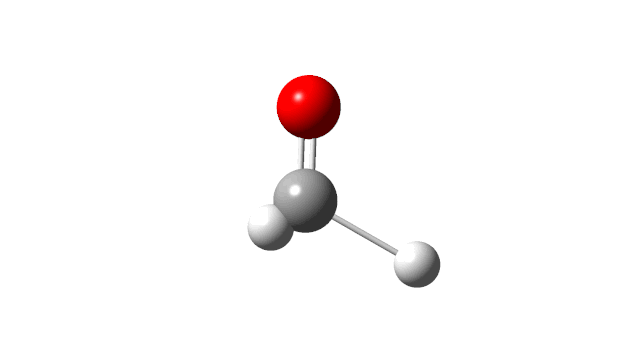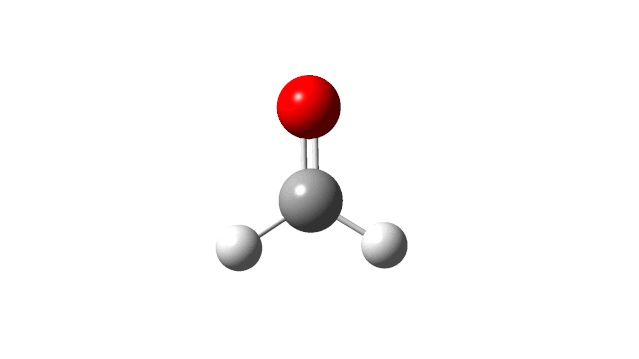
选择相应的频率,并点击’Start Animation’按钮,就可以看到振动模式,甲醛分子的6个振动模式如下:
(1) CH2 wag
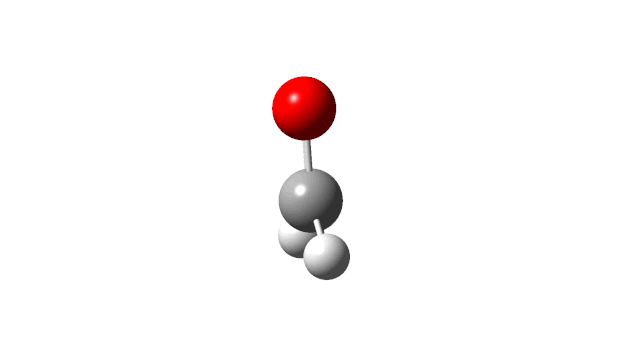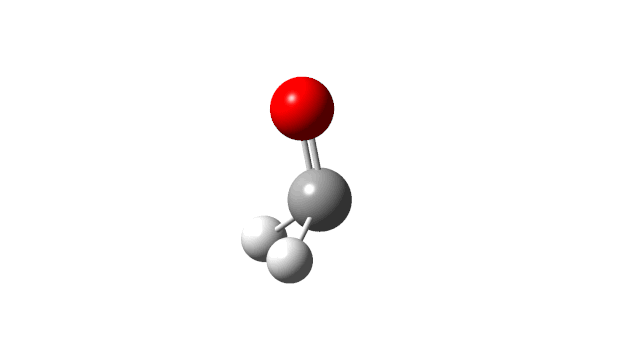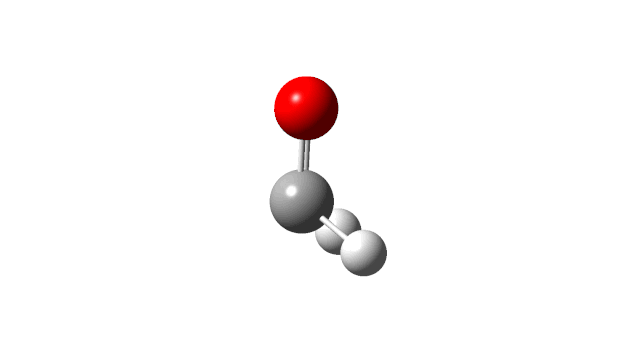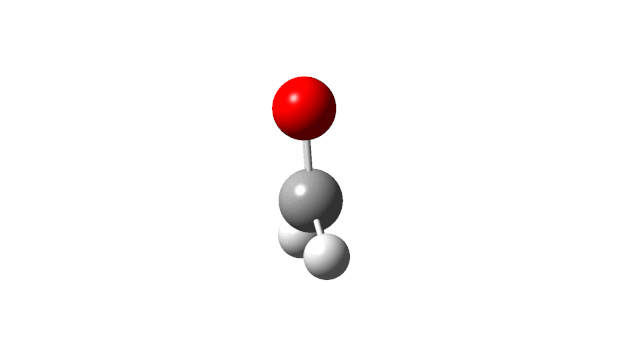
(2) CH2 rock
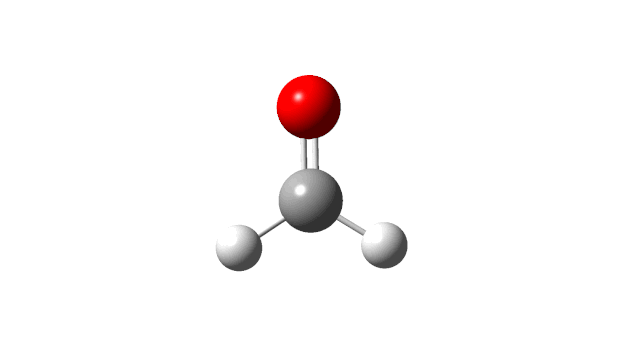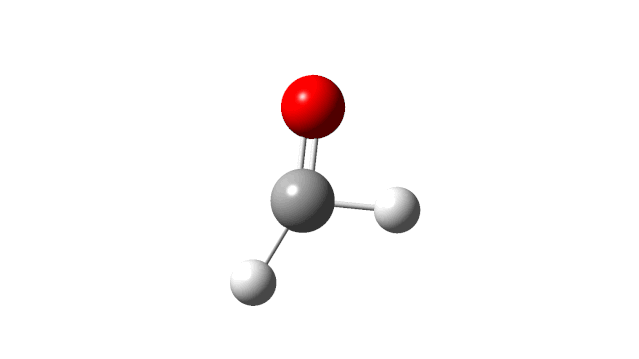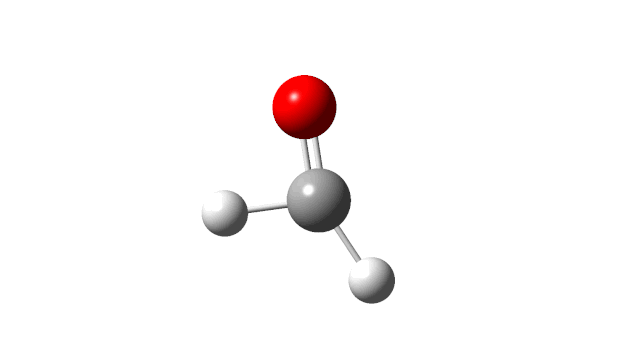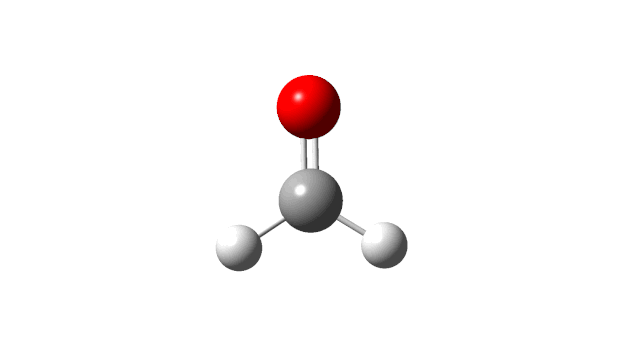
(3) CH2 scissors
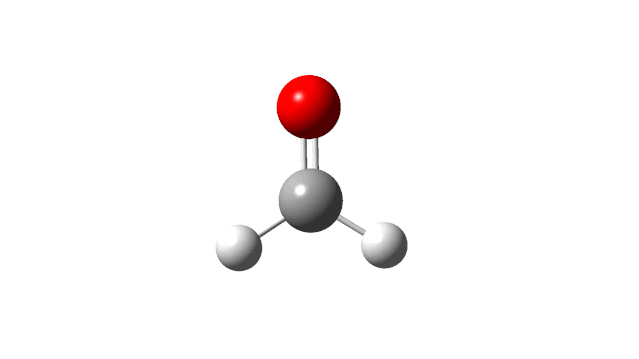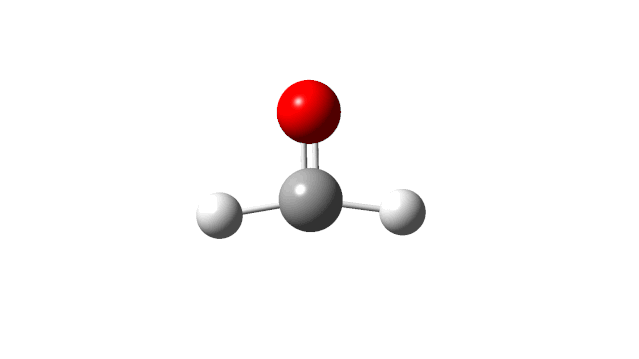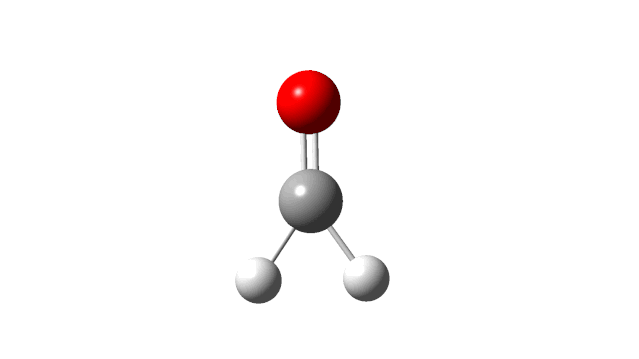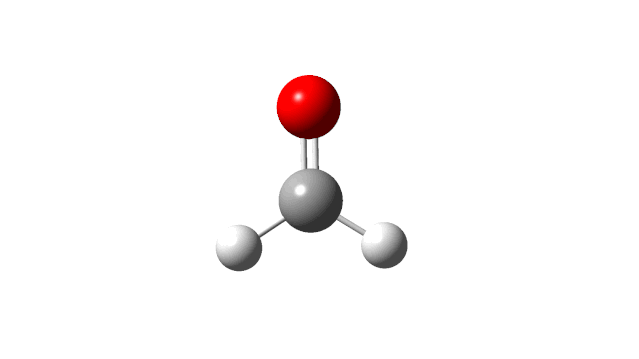
(4) C=O stretch
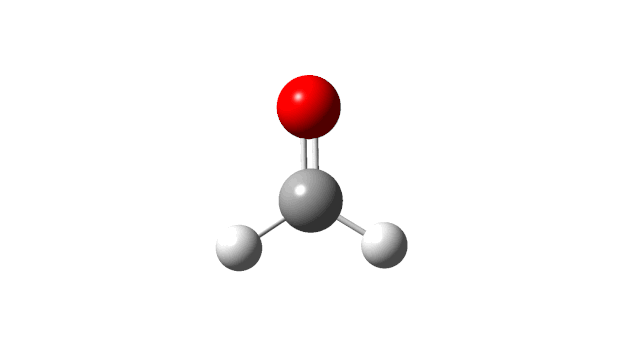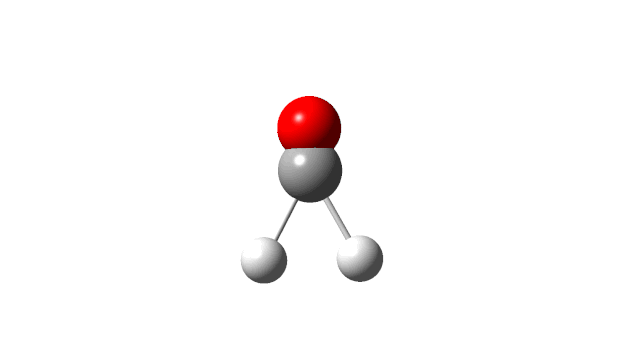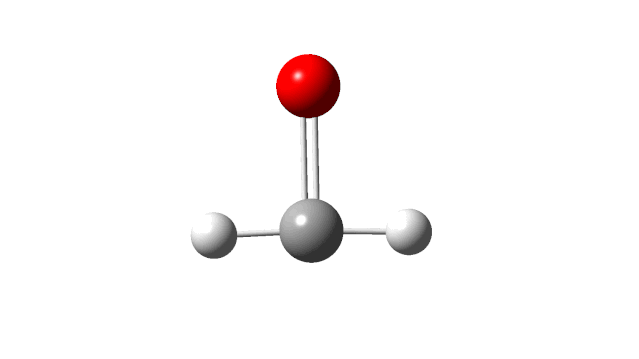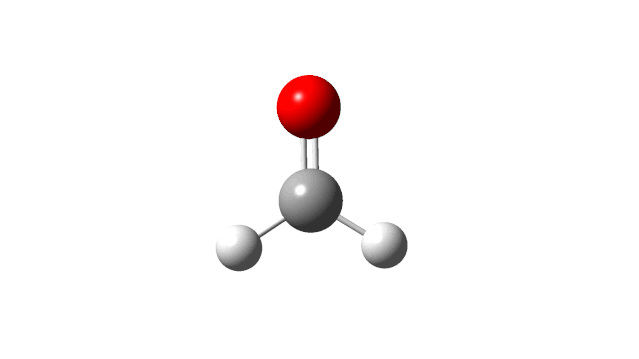
(5) C−H symmetric stretch
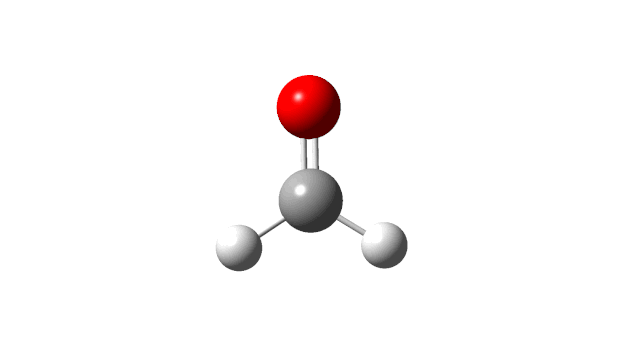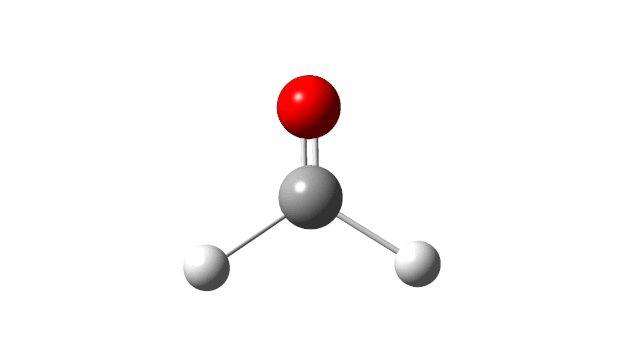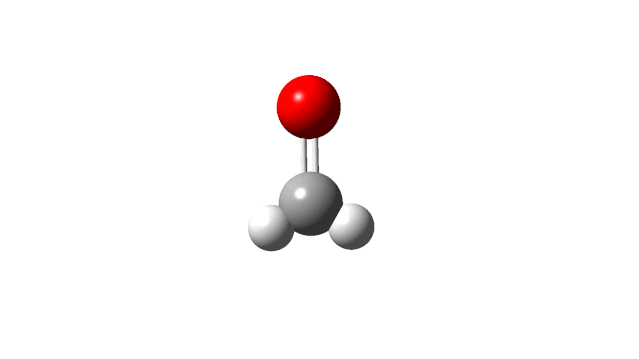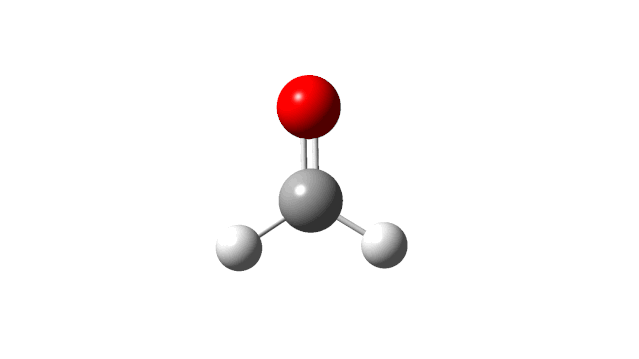
(6) C−H asymmetric stretch
三、频率校正因子
由于计算方法本身的误差以及谐振近似的使用,使得理论计算的红外频率一般无法与实验结果(基频)对上。例如Hartree-Fock方法由于没有电子相关效应,通常会高估10%~12%。在实际应用中,常常将HF计算值乘上一个校正因子0.8928。对DFT方法,所得的结果就准确多了,例如Bauschlicher和Partridge在B3LYP/6-311+G(3df,2p)水平下拟合的校正因子为0.989,比较接近1了。在文献中,针对不同的计算水平,前人已经总结了大量的校正因子,需要注意的是不同方法和不同基组的校正因子都不同,即使同样的方法基组,在不同的文献中由于拟合的数据和方式的不同,校正因子也可能不同。下列这篇文献中有常见的计算水平的校正因子可供需要的小伙伴参考:
J. Phys. Chem. A 2015, 119, 1701−1714
对上述甲醛分子,我们采用这篇文献中的校正因子0.9671,在Vibrations界面的下方有Scale frequencies?对话框,选择Specify,输入校正因子,就会在上方出现Scaled Freq一列:
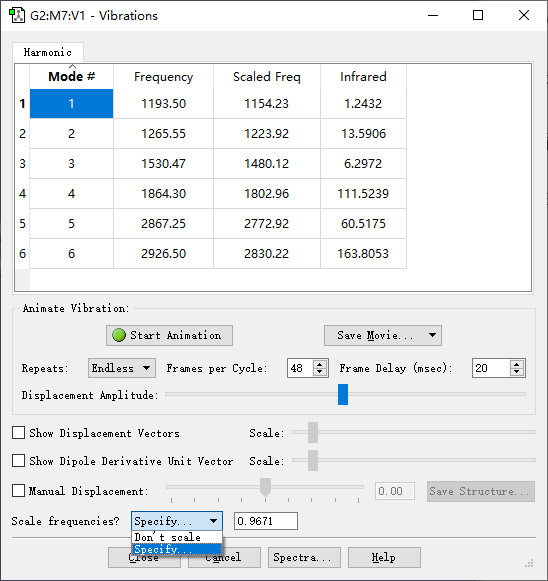
与实验值对比如下:
calculated | scaled | observed |
|---|---|---|
1194 | 1154 | 1167 |
1266 | 1224 | 1280 |
1530 | 1480 | 1503 |
1864 | 1803 | 1744 |
2867 | 2773 | 2780 |
2927 | 2830 | 2874 |
四、谱图的绘制
点击Vibrations界面下方的Spectra按钮,就会出现谱图:
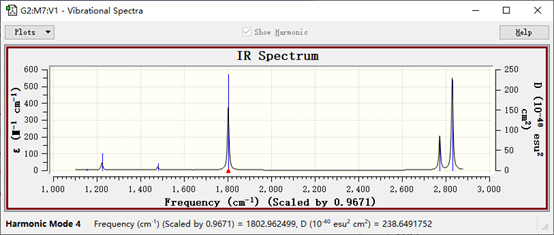
实验化学家绘制的红外光谱通常纵坐标是透射率,横坐标是从大到小的频率,因此我们一般将图的X和Y轴都颠倒一下,就符合我们常见的红外谱图的样子了。在图像上右击或者点击左上角的Plots按钮,选择Properties,出现如下设置界面:
各项含义都比较明确,在此就不细说了。最后可以点击Plots下拉菜单中的Save Picture选项,存成图片:
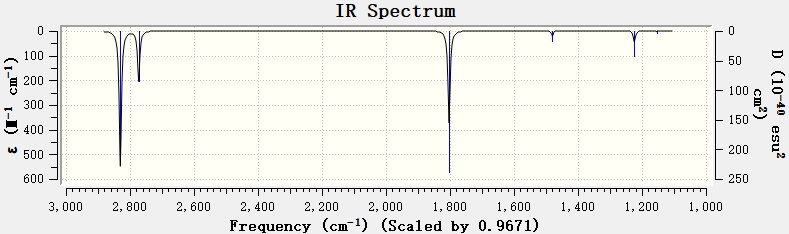
也可以不使用GaussView绘图,而是从输出文件中获取频率和相应的红外强度进行绘图。
Harmonic frequencies (cm**-1), IR intensities (KM/Mole), Raman scattering
activities (A**4/AMU), depolarization ratios for plane and unpolarized
incident light, reduced masses (AMU), force constants (mDyne/A),
and normal coordinates:
1 2 3
A A A
Frequencies -- 1193.4971 1265.5519 1530.4688
Red. masses -- 1.3759 1.3515 1.0988
Frc consts -- 1.1547 1.2754 1.5164
IR Inten -- 1.2432 13.5906 6.2972
Atom AN X Y Z X Y Z X Y Z
1 6 0.00 -0.00 -0.18 0.00 0.15 -0.00 0.00 -0.00 -0.00
2 8 0.00 0.00 0.04 -0.00 -0.08 0.00 -0.08 0.00 -0.00
3 1 -0.00 0.00 0.70 -0.65 -0.25 0.00 0.61 0.36 0.00
4 1 -0.00 -0.00 0.70 0.65 -0.25 -0.00 0.61 -0.36 0.00
4 5 6
A A A
Frequencies -- 1864.2979 2867.2495 2926.5048
Red. masses -- 7.6031 1.0466 1.1208
Frc consts -- 15.5694 5.0697 5.6554
IR Inten -- 111.5239 60.5175 163.8053
Atom AN X Y Z X Y Z X Y Z
1 6 0.60 -0.00 -0.00 0.06 -0.00 0.00 -0.00 -0.10 -0.00
2 8 -0.42 -0.00 0.00 0.00 0.00 0.00 0.00 0.00 -0.00
3 1 -0.18 -0.45 -0.00 -0.36 0.61 -0.00 -0.37 0.60 -0.00
4 1 -0.18 0.45 -0.00 -0.36 -0.61 -0.00 0.37 0.60 0.00对比较小的分子,高斯的输出文件中还会绘制一个谱图:
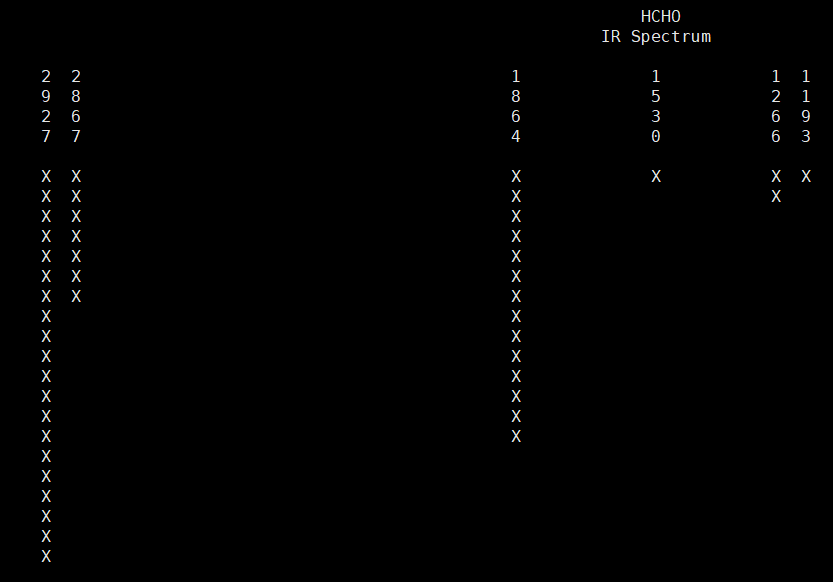
应该是程序员闲得无(dan)聊(teng)写的。自己根据数据绘制谱图时需要注意的是理论计算得到的谱图是孤立的线,作图时需要使用展宽技术,这方面的原理和操作,我们以后介绍。
参考文献:
[1] 彭笑刚,物理化学讲义
[2] 邢其毅等,基础有机化学(第四版)
[3] 范康年,谱学导论(第二版)
[4] J. B. Foresman and Æ Frisch, Exploring Chemistry with Electronic Structure Methods, 3rd ed
[5] G. Herzberg, Molecular Spectra and Molecular Structure II. Infrared and Raman Spectra of Polyatomic Molecules