单分子标签技术(Unique Molecular Identifier, UMI)被广泛应用在极高灵敏度的NGS检测中,尤其是目前炙手可热的循环肿瘤DNA (ctDNA) 检测。ctDNA作为一种非侵入性的肿瘤生物标志物,以其极高的灵敏度,可用于癌症早筛早检,治疗反应的实时监控等。因此,大量的研究工作围绕ctDNA而展开。然而,由于传统NGS检测灵敏度受限于PCR和测序的错误率(~1%),必须通过UMI来保证ctDNA检测的高灵敏度和特异性。
UMI技术的原理是在PCR扩增前给每一条原始DNA加上一段特有的短标签序列,在建库和测序完成之后可以根据标签序列和比对位置回溯到原始DNA,通过比对来源于同一DNA的多条序列的共同序列(consensus)来甄别突变是真实的还是引入的随机错误。当需要检测的目标突变的变异丰度低于1%的时候,UMI的使用可以大幅提升变异检测的准确性。

然而,以fgbio为代表的UMI consensus软件使用的统计模型比较粗糙,对不同随机过程的统计模型缺少严谨的预估,反而依赖复杂的流程和用户经验参数设置来优化结果,因此影响了UMI consensus 结果的准确性,也限制了后续变异检测的灵敏度。另一方面,为了提高更低丰度变异检测的灵敏度,很多UMI应用已经将测序深度提高到了2万甚至5万,而为了检测更多癌症类型,panel覆盖基因数目也越来越多。因此,对应的生信流程需要处理的数据量也随之大幅增加,对UMI流程的执行效率提出了更高的要求。
为了解决上述的问题,Sentieon和客户反复调研之后,在2019年11月发布了UMI处理模块,旨在为业界提供优质、准确、高效的解决方案。Sentieon UMI处理流程极度精简,参数简单,运算高效,且依然坚持了Sentieon产品一贯的数学和工程的严谨。Sentieon UMI consensus 模块通过严谨的统计模型重构了建库和测序过程中的各类错误对结果的影响,通过机器学习自动计算样本数据的多维度参数,更加准确的执行合并同组序列。同时,Sentieon UMI流程最大限度的保留和充分利用了所有输入数据的信息,为后续变异检测提供了尽可能全面和准确的统计信息。
模拟数据测试
我们将通过模拟数据和稀释样本数据来验证Sentieon UMI流程的准确性。首先,我们使用模拟数据来对比consensus molecule和原始read,检验consensus统计模型的准确性。然后,通过稀释样本来检验UMI流程对变异检测的影响。
首先,我们随机生成了1百万条DNA片段,片段长度从100到500bp不等。这些片段将是后续验证UMI consensus所对应的真值。然后,我们模拟了PCR扩增和测序两个步骤,生成了约2千万个序列,PCR扩增模拟了8个PCR循环(cycle),在每组扩增出的序列中随机选取3个序列,在此过程中按照指定的错误率随机引入了SNP和indel的错误。测序过程使用ART来模拟Hiseq2500的测序过程,通过测序仪真实的错误模型来生成测出序列。针对双端UMI,我们还引入了单链DNA错误。
模拟结果分别经过Sentieon和fgbio流程执行合并(consensus)处理。然后,我们将两个流程的处理结果consensus molecule和原始序列比对,以检验结果的正确性。
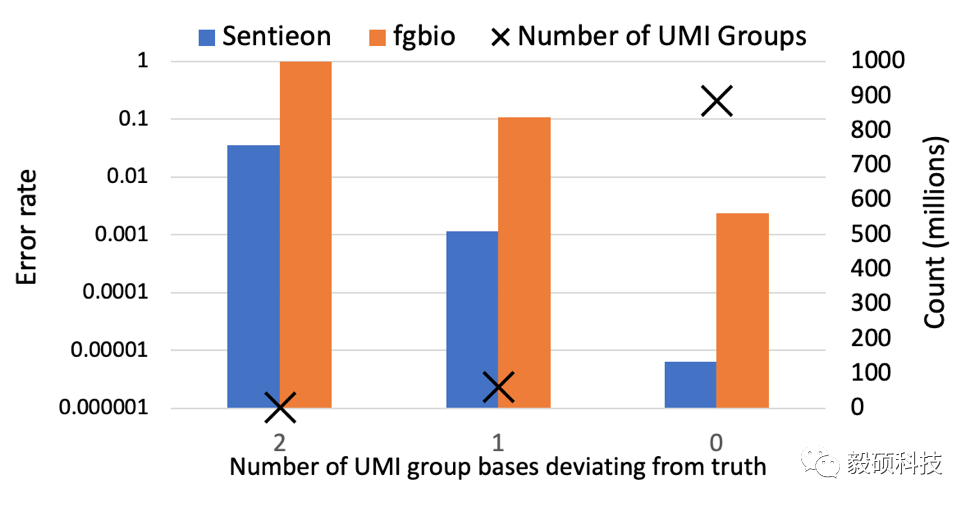
因为在PCR过程中我们随机抽取了三个序列,所以,每个位点将从三个bases合并成一个consensus base。为了更仔细的对结果进行评估,我们把每个位点上的pile up分成三类:较为常见的情况下3个碱基全部一致(如AAA), 2个一致(如AAC),和较为罕见的3个都不同的情况(如ACG)。从上图的结果中可以看出,在所有这3种情况下,Sentieon UMI consensus流程的错误率都比fgbio降低了两个数量级或以上。这里需要注意的是,即使在输入序列的三个碱基全部一致的情况下,Sentieon和fgbio流程均还有错误。这是因为虽然一个原始碱基,例如C,因为PCR和测序错误变成三个完全与C不同的,例如AAA,的可能性非常小,但是在大量的模拟PCR扩增数据情况下,我们依然可以看到这样的情况发生。
fgbio的错误率高主要体现在缺少对PCR扩增错误率的正确预估,以及放弃了将序列质量较低的位点,将它们直接标记为N,因此增大了错误率。例如,在上图中,在ACG和AAC的两种情况下(左侧和中间两个bar),fgbio基本上完全放弃了这些位点,但是通过严格的统计模型预估,我们将这些位点的错误率分别降低到了5%和1‰。
稀释样本测试
测试真集来自于2018年Qiagen发表的相关文章(doi: 10.1093/bioinformatics/bty790)中NA12878和NA24385细胞系DNA的实验室混合 (in vitro mix)。测序深度在UMI consensus之前是59000x,consensus之后大约7000x,目标突变的变异丰度约为0.5%。数据分别经过Sentieon UMI流程和fgbio流程处理之后,通过Sentieon TNscope使用相同参数进行体细胞变异检测,结果与真集比对绘制ROC曲线。

从结果可以看出,无论是SNV(左图)还是Indel(右图),Sentieon UMI流程的准确度均更高。从F1 Score来看,SNV方面Sentieon vs Fgbio是0.823 vs 0.748,Indel方面Sentieon vs Fgbio是0.742 vs 0.573。值得一提的是,Sentieon软件流程按照模块化设计,consensus之后的bam文件可以输入到各类变异检测软件,具有良好的兼容性。
速度测试
Fgbio流程冗长繁琐,完成全部UMI处理一共要8个步骤,执行效率较低。相比之下,Sentieon UMI在设计流程时,充分从易用性的角度出发,大幅精简了UMI流程,仅仅三步即可。在通过更复杂和严谨的统计模型保证准确性的前提下,Sentieon坚持了其一贯的代码高效稳定的风格,UMI 流程整体处理速度对比Fgbio提高了20倍左右(32线程工作站),极大的提升了UMI处理的整体速度,配合Sentieon的变异检测模块,为大panel高深度测序产品提供一个高效准确的分析流程框架。

设计原理
Sentieon UMI设计的核心理念是不丢弃任何原始序列包含的可用信息,经过统计模型的处理之后为每一个consensus序列的每个位点赋予一个准确的质量值,供下一步的变异检测工具参考。举例来说,在一些例如同组UMI序列数量不足,序列正反链碱基不一致,原始序列质量值过低等情况下,以fgbio为代表的工具倾向于采用剪裁丢弃序列,或者直接标记N等简单粗暴的做法。相比之下,Sentieon通过严格的统计模型,准确预估了例如单链错误率,PCR错误率,测序错误率等参数,然后对每个consensus序列提供可靠的置信度评估。这样,后续的变异检测可以获取到充分的统计信息,并相应的对检测到的可能变异做出正确的评估。
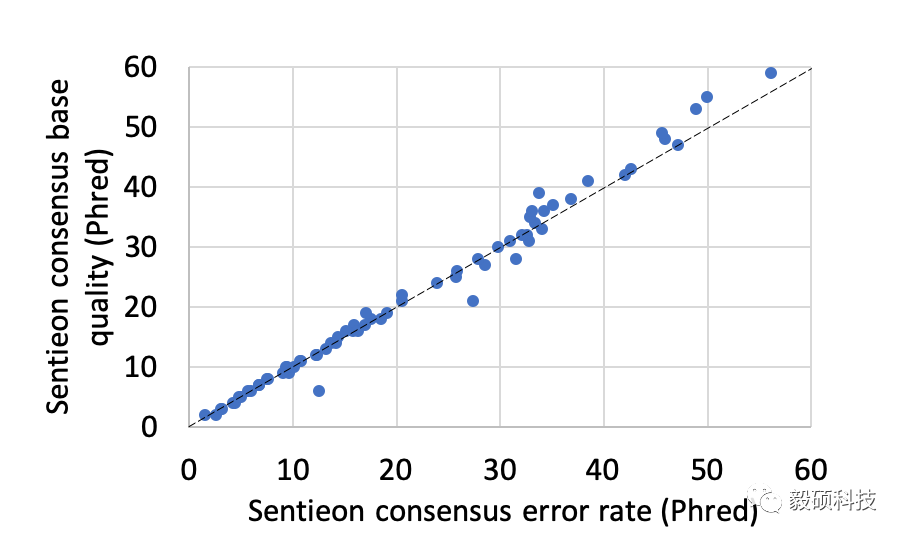
在我们的模拟数据测试中,我们分析了统计模型给出的质量值和实际的错误率之间的关系,针对所有模拟的情况,包括不同的单链、PCR和测序错误率。从上图统计中我们发现,我们给出的质量值高度准确的体现了这些统计过程的所引入的随机错误。这证明了Sentieon UMI不但在建立consensus序列上有极高的正确性,同时也能为每个consensus序列提供非常有价值的质量值。
常见问题
1. 对于特殊类型的UMI,比如说动态长度的UMI,Sentieon软件能处理吗?
目前不能直接使用,但是UMI extract开源,用户可以自己修改满足不同的UMI需求。
2. 支持双端UMI吗?
支持。软件会估算两条链不一致的错误率,并对结果做修正。
3. 流程中的统计学模型的参数需要提前训练吗?
不需要,所有参数均直接从测试样本数据中学习。
4. 在去接头的步骤中,Sentieon UMI流程是如何处理的?
Sentieon UMI并没有专门的去接头步骤,只是在处理有重叠的pair-end reads时,如果pair-end reads相互测通,则把超出的部分切除。
5. 在通过统计学模型计算出质量值以外,软件会保存原始的序列信息吗?
会的,软件会把family size等原始信息写在BAM文件的XZ tag里,供用户查询或者进行个性化的进一步过滤。
6. 序列的过滤推荐在哪一步进行?
Adapter trimming最好在UMI处理之前进行,在consensus之后软件会尽量保留所有的序列,以质量值区分可信度。所以如果后续变异检测软件能够读取并利用质量值,那么无需在变异检测之前进行过滤。如果质量值无法被利用,用户可以根据质量值和写在XZ tag里面的原始信息进行自行过滤。