导语
GUIDE ╲
对癌症样本的系统DNA测序强调了癌症基因组学的两个方面的重要性:肿瘤内异质性(ITH)和突变过程。
背景介绍
肿瘤内异质性(ITH)和突变过程可能并不总是独立的,因为不同的突变过程可能涉及到肿瘤的不同阶段或区域,但现有的研究它们的计算方法在很大程度上忽略了这种潜在的依赖性。
今天小编给大家推荐的一篇文章,提出了CloneSig计算方法,在肿瘤中从批量测序数据联合推断ITH和突变过程的关联。文章于2021年发表在《NATURE COMMUNICATIONS》杂志上,最新影响因子为14.919,题目为:CloneSig can jointly infer intra-tumor heterogeneity and mutational signature activity in bulk tumor sequencing data.

数据介绍
TCGA:从GDC获取CNV和CNA数据(33种癌症)
PCAWG队列:从ICGC下载了SNV和拷贝数calls
DREAM:syn2813581
PhylogicSim500 and SimClone1000数据集
结果解析
01
CloneSig联合估计ITH和突变过程
作者提出了CloneSig,一种从肿瘤样本的整体基因组测序数据中联合推断ITH和估计不同克隆中活跃的突变过程的方法。CloneSig背后的基本原理如图1所示,显示了WES在肉瘤(TCGA患者TCGA-3BA9HI)中检测到的所有SNVs沿两个轴的散点图。
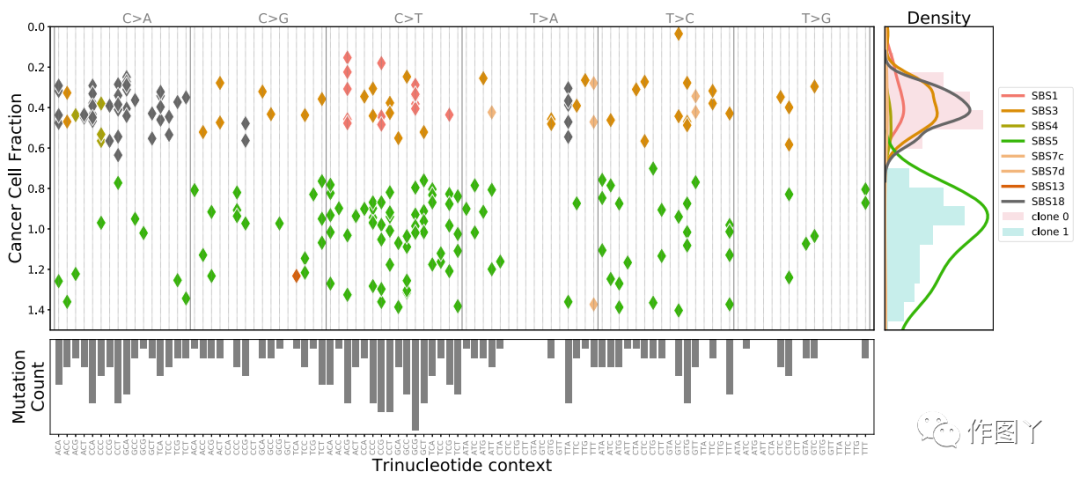
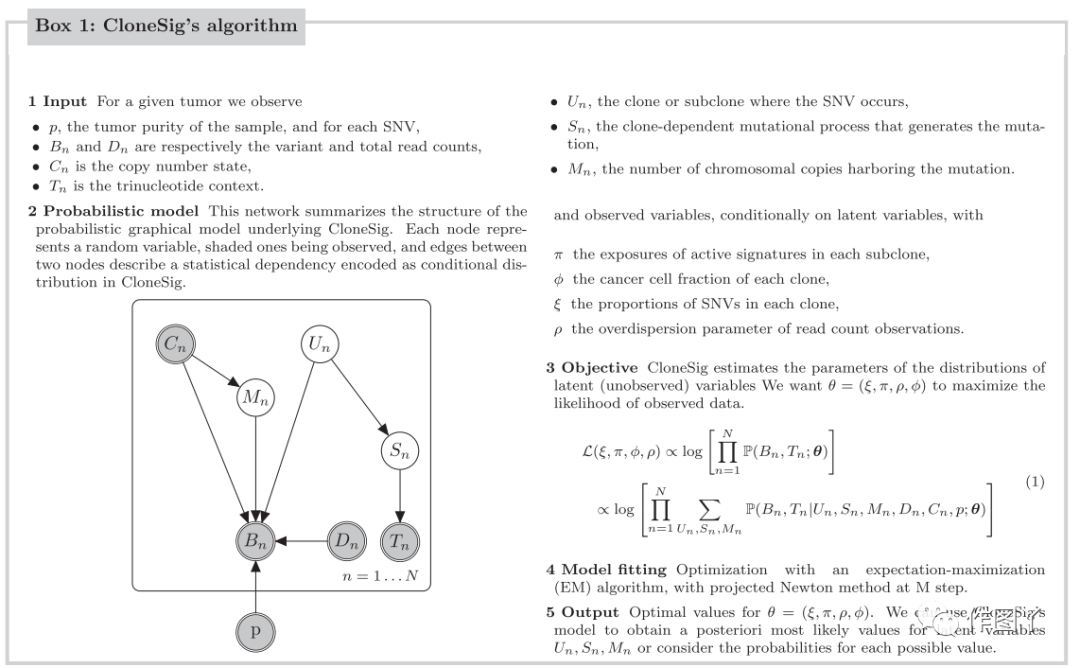
图1
02
亚克隆重建的性能
首先评估了CloneSig在模拟数据上正确重建肿瘤亚克隆组织的能力,使用四种不同的模拟数据:(1)DREAM数据集,由5个不同测序深度的模拟WGS肿瘤组成,(2)PhylogicSim500,(3)SimClone1000,(4)CloneSigSim,根据CloneSig背后的概率图形模型来模拟数据。
图2总结了CloneSig和其他ITH重建方法在“constant”和“varying”下的亚克隆重建性能。每个雷达图都显示了每种方法在一组模拟数据上达到的平均得分(1B、1C、2A和2C)。在“constant”场景下,看到CloneSig在所有分数和所有模拟器上都与最好的ITH方法(PhylogicNDT、DPClust和Ccube)相同或更好,而CciClone、TrackSig和TraccSigFreq的整体性能较差。

图2
图3总结了PhylogicSim500模拟(在“constant”和“varying”设置中,分别对应于“constant”和“varying” signature设置).
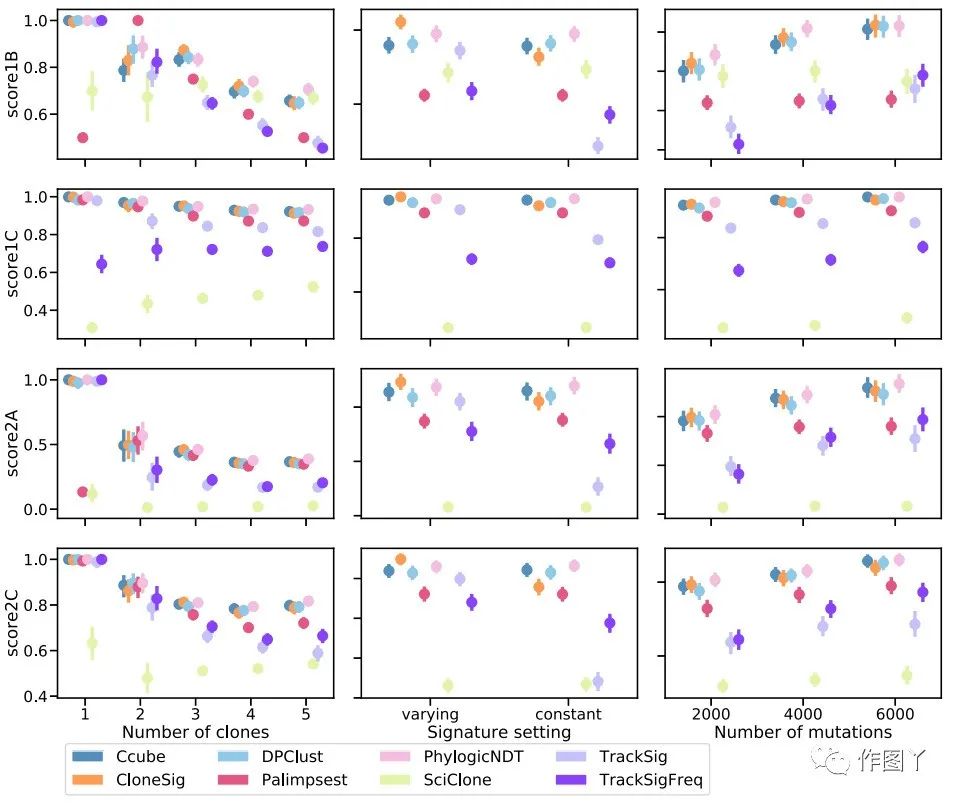
图3
为了进一步说明signature变化和检测克隆的能力之间的相互作用,现在用恰好有两个克隆的模拟来测试CloneSig、TrackSig、TrackSigFreq和重文本,并且改变了克隆在CCF方面的差异和在突变过程方面(根据两种类型突变谱之间的余弦距离来量化)。
图4显示了CloneSig作为这两个参数对克隆和亚克隆突变进行正确分类的ROC曲线下的面积(AUC)。我们看到,随着突变类型谱之间距离的增加,AUC增加,因为克隆之间的CCF差异恒定。
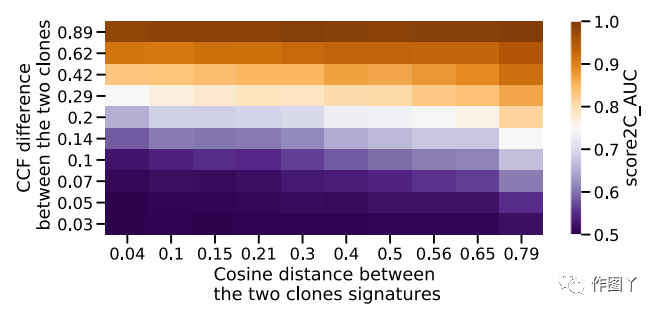
图4
03
signature活性反卷积的性能
图5显示了在PhylogicSim500数据集上根据不同指标使用的不同方法的性能。对于Score_sig_1A和Score_sig_1B,CloneSig、TrackSig和trackSigFreq具有重写本和解构信号,除非突变数最小,在这种情况下,所有方法的行为相似。对于Score_sig_1C,CloneSig、TrackSig和TraccSigFreq显示出最佳的AUC,以检测所有场景中的当前签名。
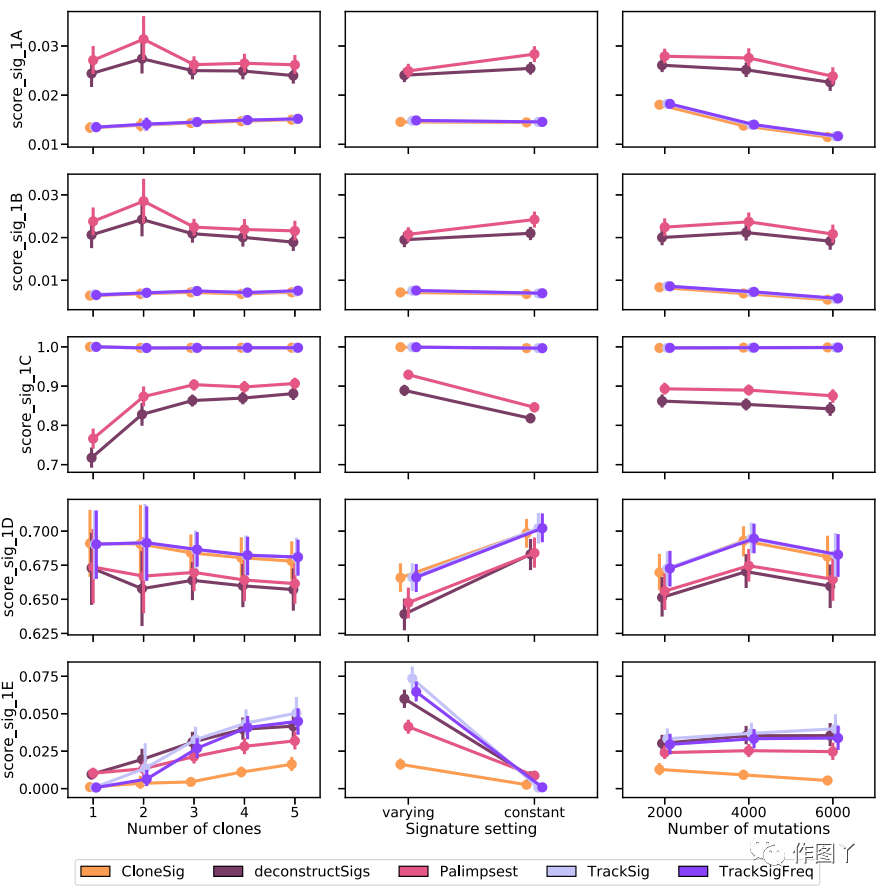
图5
04
特征变化的泛癌分析
图6显示了在TCGA队列中发现的signature变化的整体分析。对于每个癌症类型至少100名患者,它显示了发现signature更改的样本比例,以及对最大亚克隆中相比克隆突变每个个体签名增加或减少的样本比例的可视化总结。此外,每个条的厚度表示每个signature的中位数变化。只保留了在每个队列样本中超过10%中发现变异的签名。

图6
小编总结
CloneSig在ITH推断和检测突变过程方面优于当前的方法。CloneSig应用于来自TCGA的8951个肿瘤的全外显子组测序数据的大队列,以及来自全基因组全癌症分析计划中的2632个全癌症肿瘤样本的泛基因组测序数据集上,获得的结果总体上与之前的研究一致。
近来模型构建的文章层出不穷,本文作者别出心裁的关注了肿瘤内异质性(ITH)和突变过程这一生物学问题,利用多类数据集建模,效果也是十分的好,小伙伴们在学习过程中也要多多思考要针对的生物学问题,然后再进行研究,才能达到好的效果哦!