题目:构造苯、丁二烯分子,在 B3LYP-D3/cc-pVDZ水平优化几何构型。在CASSCF/cc-pVTZ水平使用对称性计算这些分子的基态(ground states)电子结构,活性空间只包括所有价层π轨道。
参考解答
(1)构造苯\丁二烯分子,在 B3LYP-D3/cc-pVDZ水平优化几何构型。输入文件略,优化后的分子坐标见附录。
(2)在CASSCF/cc-pVTZ水平使用对称性计算这些分子的基态。
使用BDF:
① 使用iCAS方法寻找活性空间初始猜测轨道。苯分子的输入文件如下:
$compass title benzene basis cc-pvtz geometry file=benzene.xyz end geometry group D(2h) $end$xuanyuan
$end$scf
guess
huckel
RHF
charge
0
spin
1
atomorb
molden
$end%cp BDF_WORKDIR/BDFTASK.scforb BDF_WORKDIR/BDFTASK.inporb
$expandmo
vcmo
minbas
6
1C|2P0
2C|2P0
3C|2P0
4C|2P0
5C|2P0
6C|2P0
end</code></pre></div></div><p>本次计算苯用的对称性为D(2h),其8个不可约表示在BDF中的顺序依次为Ag,B1g,B3g,B2g,Au,B1u,B3u,B2u。expandmo模块计算结束给出的轨道信息可用于CASSCF计算的输入文件:</p><figure class=""><div class="rno-markdown-img-url" style="text-align:center"><div class="rno-markdown-img-url-inner" style="width:80.87%"><div style="width:100%"><img src="https://cdn.static.attains.cn/app/developer-bbs/upload/1723340177075868243.png" /></div></div></div></figure><p>② 苯的活性空间为CAS(6,6),丁二烯的活性空间为CAS(4,4),苯的输入文件如下:</p><div class="rno-markdown-code"><div class="rno-markdown-code-toolbar"><div class="rno-markdown-code-toolbar-info"><div class="rno-markdown-code-toolbar-item is-type"><span class="is-m-hidden">代码语言:</span>javascript</div></div><div class="rno-markdown-code-toolbar-opt"><div class="rno-markdown-code-toolbar-copy"><i class="icon-copy"></i><span class="is-m-hidden">复制</span></div></div></div><div class="developer-code-block"><pre class="prism-token token line-numbers language-javascript"><code class="language-javascript" style="margin-left:0">compass
title
benzene
basis
cc-pvtz
geometry
file=benzene.xyz
end geometry
saorb
group
D(2h)
$end$xuanyuan
$end%cp BDF_WORKDIR/BDFTASK.exporb BDF_WORKDIR/BDFTASK.inporb
$mcscf
guess
read #读取上一步expandmo计算得到的轨道.exporb作为初猜
actel
6
close
6 3 0 0 0 0 5 4
active
0 0 1 2 1 2 0 0
spin
1
molden
symmetry
1
iprtmo
1
$end
将CASSCF计算得到的molden文件用软件Multiwfn打开,检查6个π轨道是否被选进活性空间。CAS(6,6)活性空间的6个轨道91,109,110,133,151,152依次如图1所示,可看出都是我们想要的轨道。(BDF的CASSCF轨道是按照不可约表示排序的,每个不可约表示内部按照双占、活性、虚轨道顺序排序。)

图1 苯CAS(6,6)活性空间内的轨道
经过类似的步骤,可以得到丁二烯分子的4条CASSCF活性轨道分别为71,72,173,174,如图2所示

图2 丁二烯CAS(4,4)活性空间内的轨道
使用BDF得到的苯和丁二烯的能量如表1所示:
表1 使用BDF得到的RHF与CASSCF能量(in Hartree)

使用ORCA:
我们将分别使用HF和MP2的轨道作为CASSCF计算的初始猜测。
① 使用HF轨道作为初猜
苯的RHF计算的输入文件:
!RHF cc-pvtz UseSym pal4
*xyzfile 0 1 benzene.xyz
ORCA默认不使用对称性,使用对称性的计算需加!UseSym。将计算产生的.gbw文件转换成molden文件,寻找要选进活性空间的轨道。经过查看,苯的π轨道为17,20-23,30,如图3所示。丁二烯的π轨道为14-16,22,如图4所示(轨道编号从1开始)。
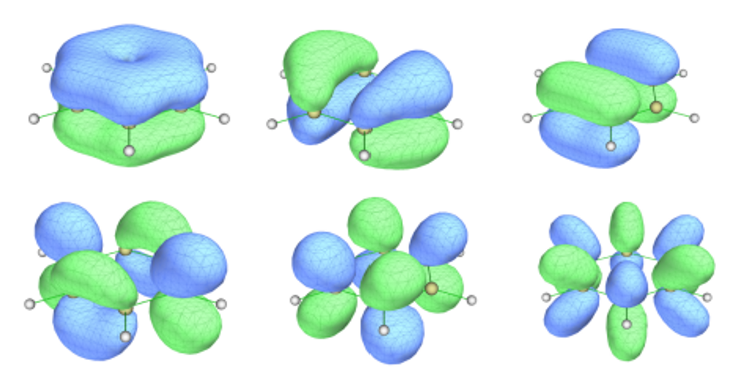
图3 苯要选进活性空间的轨道
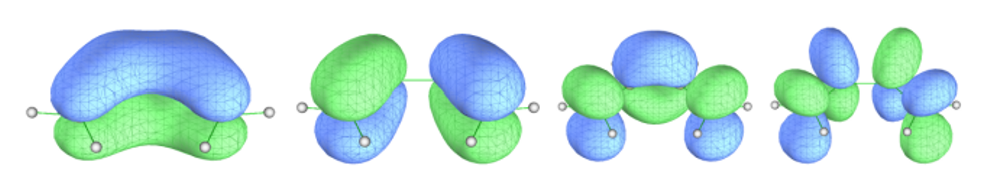
图4 丁二烯要选进活性空间的轨道
因此,使用RHF轨道作为CASSCF初始猜测,需要交换轨道顺序。对于苯需要交换轨道17和19,轨道24和30;对于丁二烯需要交换轨道17和22。注意,在ORCA中轨道是从0开始的,所以上述数字都需要减1。
苯的CAS(6,6)计算的输入文件如下:
!cc-pvtz UseSym pal8
!moread
%moinp "benzene-rhf.gbw"
%scf
rotate {16,18,90} {23,29,90} end
end
%casscf
nel 6
norb 6
mult 1
irrep 0
end
*xyzfile 0 1 benzene.xyz最后可以将CASSCF计算产生的.gbw文件转换成.molden文件,检查活性轨道是否正确。以上计算得到的能量数据如表2所示:
表2 使用ORCA得到的RHF与CASSCF能量(in Hartree)

② 使用MP2的自然轨道作为初猜时:
构造苯分子MP2自然轨道的计算文件为:
!MP2 cc-pvtz NoFrozenCore UseSym pal4
%mp2 Density unrelaxed
NatOrbs true
end
*xyzfile 0 1 benzene.xyz将计算产生的.mp2nat文件转换成molden文件,可以看到苯的π轨道为19-24(轨道顺序从1开始),如图5所示;丁二烯的π轨道为14-17,如图6所示。

图5 苯的初始猜测活性轨道

图6 丁二烯的初始猜测活性轨道
因此,使用MP2自然轨道作为CASSCF计算的初始猜测,对于这两个体系都不需要调整轨道。
苯的CAS(6,6)计算的输入文件:
!cc-pvtz UseSym pal8
!moread
%moinp "benzene-mp2.mp2nat"
%casscf
nel 6
norb 6
mult 1
irrep 0
end
*xyzfile 0 1 benzene.xyz得到的结果和BDF完全一致。
表3 使用ORCA得到的RHF、MP2与CASSCF能量(in Hartree)

附录
benzene.xyz 单位:angstrom
C -1.398696106140 0.000000000000 0.000000000000
C -0.699348053043 1.211306360083 0.000000000000
C 1.398696106140 0.000000000000 0.000000000000
C 0.699348053043 1.211306360083 0.000000000000
C 0.699348053043 -1.211306360083 0.000000000000
C -0.699348053043 -1.211306360083 0.000000000000
H -2.491140018813 0.000000000000 0.000000000000
H -1.245570009380 2.157390540673 0.000000000000
H 2.491140018813 0.000000000000 0.000000000000
H 1.245570009380 2.157390540673 0.000000000000
H 1.245570009380 -2.157390540673 0.000000000000
H -1.245570009380 -2.157390540673 0.000000000000butadiene.xyz 单位:angstrom
C -1.544236423239 0.000000000000 -0.505683451786
C -0.735894131124 0.000000000000 0.565591779838
C 0.735894131124 0.000000000000 0.565591779838
C 1.544236423239 0.000000000000 -0.505683451786
H -2.630182215978 0.000000000000 -0.389568240937
H -1.198520606853 0.000000000000 1.558886145620
H 1.198520606853 0.000000000000 1.558886145620
H 2.630182215978 0.000000000000 -0.389568240937
H -1.157435943160 0.000000000000 -1.528767873368
H 1.157435943160 0.000000000000 -1.528767873368