不能出门的日子,除了吃吃睡睡,你还能做什么呢?快快趁着这个机会好好修炼啊。
我们先来认识一下GSEA。GSEA(Gene Set Enrichment Analysis,基因集富集分析)是一个计算方法,用来确定某个基因集在两个生物学状态中(疾病正常组,或者处理1和处理2等)是否具有显著的一致性差异。一般通过网站下载安装后使用(https://www.gsea-msigdb.org/gsea/index.jsp)。
但是!!!你有没有遇到过这种情况呢?
试一试这个R包吧。iGSEA(Integrative Gene Set Enrichment Analysis Approaches;v1.2,2017-05-07)就是基于GSEA方法整合多个GSEA研究结果的一个混合策略。它提供三种方法:基于固定效应模型的iGSEA-FE,基于随机效应模型的iGSEA-RE,以及用于整合多个基因集富集研究的iGSEA-AT。适用于分类表型和连续表型。最终结果为基因集FDR校正后的显著性Q值。
下面我们就来认识一下它。
##安装并导入
install.packages(“iGSEA”)
library(iGSEA)
##查看帮助
??iGSEA##主要功能函数igsea.test的几个参数,其他几个参数默认即可。
gel:基因表达水平的数值矩阵(行-基因,列-样本)。
pheno:表型的数值向量。
ssize:每个研究中样本数量的数值向量。
gind:基因是否包括在研究中的0-1矩阵(1-包含,行-基因,列-研究)。
gsind :基因是否包括在基因集中的0-1矩阵(1-包含,行-基因,列-基因集)。
vtype:表型类型,“binary”或“continuous”。
我们来看看表达样本和样本标签文件格式。


1.特定基因集在两个生物学状态中是否具有显著的一致性差异
set.seed(1234)
expr=read.table("expr.txt",as.is=T,header=T,sep="\t",row.names=1) #48样本,50基因
expr=as.matrix(expr)
condition=read.table("condition.txt",as.is=T,header=T,sep="\t",row.names=1) #48样本标签,1-病,0-常
sampleNum=c(24,24) #病常样本数
geneInSample=matrix(rep(1, 50*2), 50) #两种状态都包含所有基因
##注意每个基因应该至少包含在一种状态中
geneInSet=matrix(0, 50, 1);geneInSet[1:20, 1]=1 #包含在特定基因集中的基因设置为1
igsea.test(expr,condition[,],sampleNum,geneInSample,geneInSet) #输出显著性Q值,一般认为FDR<0.05为显著差异。如果有一种状态不包含所有基因,
geneInSample=matrix(rep(1, 50*2), 50);geneInSample[7:15,1]=0 #某种状态不包含所有基因
igsea.test(expr,condition[,],sampleNum,geneInSample,geneInSet)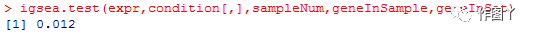
结果显示某个基因集在癌常对照中具有显著的一致性差异。
2.一次可输入多个基因集
set.seed(1234)
expr=read.table("expr.txt",as.is=T,header=T,sep="\t",row.names=1) #48样本,50基因
expr=as.matrix(expr)
condition=rep(c(1,0,1,0),c(10,10,17,11)) #48样本标签(设置10病,10常,17病,11常)
sampleNum=c(10,10,17,11) #病常样本数
geneInSample=matrix(rep(1, 50*4), 50) #四种状态都包含所有基因
geneInSet=matrix(0, 50, 2) #两个特定基因集
geneInSet[1:20, 1]=1 #包含在特定基因集1中的基因,第一列设置为1
geneInSet[38:47, 2]=1 #包含在特定基因集2中的基因,第二列设置为1
igsea.test(expr,condition,sampleNum,geneInSample,geneInSet) 
得到两个基因集的一致性显著Q值。
小编总结
GSEA网站打不开或者不方便Download应用程序,又或者我只想看看我的基因集在癌常状态中是否显著差异,那你可要试试今天的iGSEA。只要输入你的表达样本,敲两行代码就可以得到基因集的一致性显著Q值,是不是很方便快捷呢?