无论是在进行全基因组关联研究(GWAS)还是孟德尔随机化研究(MR)中,我们都需要考虑SNP间的连锁不平衡(LD)信息,这里小陈给大家简单介绍一下用于LD分析的工具-----LDlink(https://ldlink.nci.nih.gov/?tab=home),使用这个网站时最好使用代理服务器,这样比较稳定,当然不使用代理的话,有时候也是可以使用的。
进入后,网页页面如下图所示,这时候点击LDmatrix:
我们以如下SNP为例,进行演示(注意计算LD时要保证SNP在同一条染色体上):
rs10305439
rs10305442
rs1126476
rs11963172
rs11964854
rs12204668
rs2235868
rs2268650
rs2894420
rs35887128
rs6923761
rs7765641
rs7766663
rs9296290将上述SNP输入到网页框里,如下图所示:

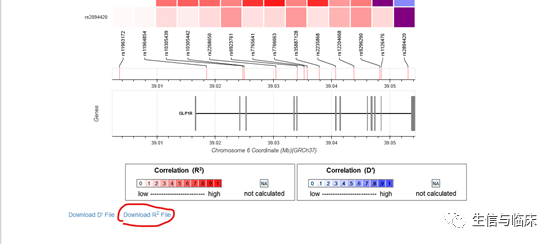
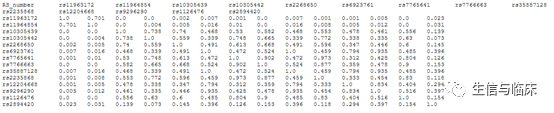
点击“Calculate”后,会返回计算结果,进入结果页面后,点击“Download R2 File”这个获取结果:
除了网页之外,LDlink还有相关的R包可供我们使用
install.packages("LDlinkR") #安装LDlinkR包
library(LDlinkR) #加载该R包
LDinfo <- LDmatrix(snps = mydata$SNP,
pop = "EUR", r2d = "r2",
token = '35deec53ae3c',
file =FALSE)这里的参数snps用于指定计算LD的那些SNP,mydata是TwoSampleMR包harmonise后的数据;参数pop是指参考基因组的人种,主要有”AFR”(非洲人), “AMR”(混合美洲人), “EAS”(东亚人), “EUR”(欧洲人)和“SAS”(南亚人)这5大类;参数r2d用于指定评估LD的指标,有两种选项"r2"和"d";参数token是一个使用身份证,大家可以自己注册申请一个;参数file指定是否保存结果。
LDinfo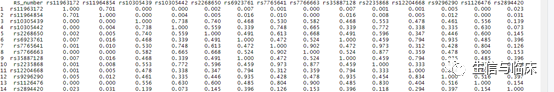
这时候我们就可以把TwoSampleMR的数据格式转化为MendelianRandomization包的那样,并矫正SNP间的相关系数:
mrinput <- mr_input(bx =mydata$beta.exposure, bxse = mydata$se.exposure,
by = mydata$beta.outcome, byse= mydata$se.outcome,
correlation =as.matrix(LDinfo[,2:ncol(LDinfo)]),
exposure = "exposure", outcome = “outcome”)这里需要注意correlation参数,它的输入要求是一个矩阵,因此我们需要对LDinfo这个结果进行转换。
关于LDlink的网页版本和R包版本的使用就先介绍到这里,希望能给大家带来帮助!接下来,小陈会继续带大家完成R语言的机器学习,敬请期待!