一个分子可以有很多的电子态,其中能量最低的称为基态(ground state),其他的态称为激发态(excited state)。通常分子通过吸收光子从基态跃迁至激发态。一般分子的基态是单重态,记为S0,部分分子的基态为三重态,记为T0。常常用Sn和Tn表示单重态和三重态第n激发态,如T1称为三重态第一激发态。每个电子态都有各自的势能面,示意图如下:
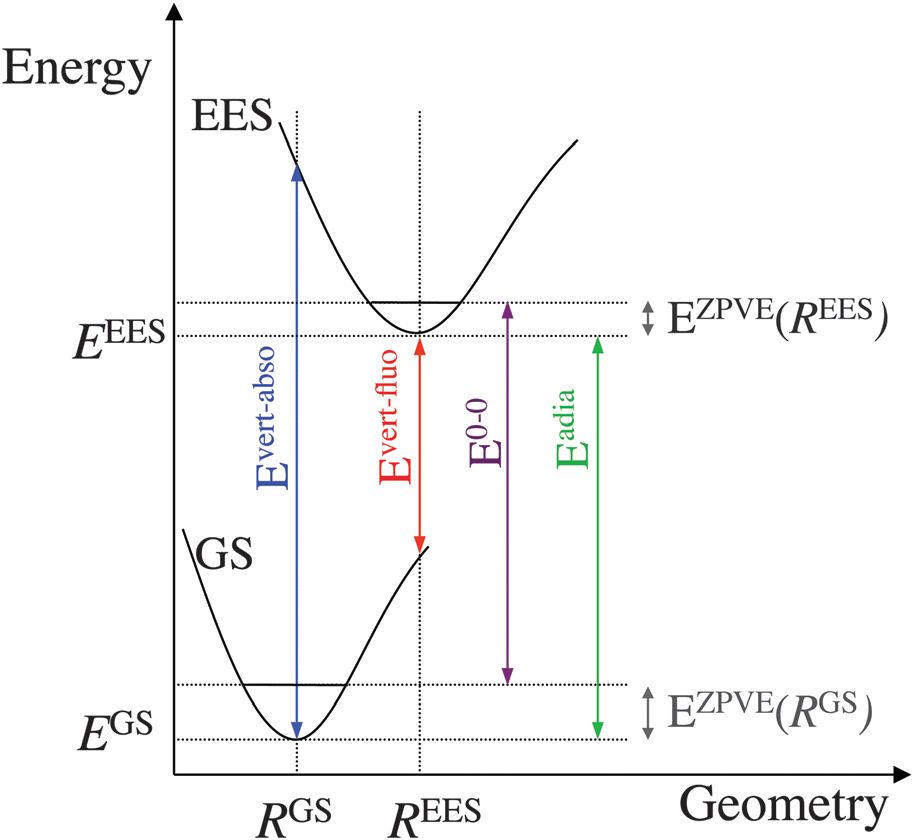
该图摘自C. Adamo, D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845,是一篇不错的关于激发态计算的入门级综述,建议大家读一读。图中EES是Electronically Excited State的缩写,R表示两个势能面的能量极小点的几何结构,对应的能量分别记为EGS和EEES,图中涉及两个电子态的零点振动能,这需要进行频率分析,本文暂不讨论。图中涉及三个重要的吸收(或激发):(1)Evert-abso,垂直吸收,图中蓝线对应的吸收。所谓垂直的含义是激发态的结构为基态的结构;(2)保持激发态能量极小点结构,到基态的发射为垂直发射,图中红线所示,当激发态为单重态时,对应的即为荧光发射Evert-fluo;(3)绝热吸收或发射,即两个势能面的极小点的能量差Eadia。
一次激发态计算只能得到该结构下的垂直吸收或发射能,例如,对基态进行几何结构优化,对优化过的结构做激发态计算,则得到图中的垂直吸收;若对激发态进行几何结构优化,并对优化过的结构做激发态计算,则得到图中的垂直发射;而要想得到绝热吸收或发射,则需要对基态和激发态都进行几何结构优化。
高斯中激发态计算的常见方法:
ZIndo:这是一种用于激发态计算的半经验方法,适用于比较大的体系。需要注意的是ZIndo方法只对部分元素有参数。
CIS:单激发组态相互作用。曾经比较流行的激发态计算方法,但是由于精度不是很高,目前已经基本被TDDFT方法取代了。
TD-HF、TD-DFT:含时的Hartree−Fock或密度泛函方法。TD-DFT是目前计算激发态的主流方法。关于激发态计算中泛函的选择,可参考卢天老师的博文:http://sobereva.com/272
EOM-CCSD:高精度的激发态计算方法,耗时也较高。
CASSCF:多组态方法,计算量随体系(活性空间)大小呈指数级增长,适合处理自由基和过渡金属等复杂情况。可以作为更高精度方法CASPT2的初始。
在激发态计算中,常常需要设置以下几个选项:
root=n
指定需要研究第n个激发态。默认值为1,即计算能量最低的激发态的相关性质。
nstates=m
计算能量最低的m个激发态,默认值为3。一般需要比感兴趣的态多算几个态,如取n+2。
triplets
求解三重态激发态。该关键词只对闭壳层单重态基态有用。
50-50
同时计算n个单重态和n个三重态。也只对闭壳层单重态基态有用。
例如TD(nstates=8,root=2,triplets)关键词表示求解8个三重态激发态,其中能量最低的第二个激发态为感兴趣的激发态。
以下以“苯在乙醇溶液中的紫外-可见光谱的理论计算”为例展示最简单的激发态的计算。
一般情况下,分子的电子能级间跃迁需要的能量约为1~20 eV,相应的波长为1230~62 nm,而紫外-可见光区的波长为200~800 nm,分子吸收紫外-可见光获得的能量可使价电子发生跃迁,得到紫外-可见吸收光谱。要从理论上模拟紫外-可见光谱只需要对体系做激发态计算即可。而激发态计算得到的是一系列激发态的能量及其振子强度,对应的谱图将是一系列孤立的线。实际的做法是将每条谱线进行展宽,最后将所有的峰加和,得到最终的紫外-可见吸收光谱。下图为卢天老师博文《使用Multiwfn绘制红外、拉曼、UV-Vis、ECD、VCD和ROA光谱图》(http://sobereva.com/224)中的示意,对此进行了非常好的解释。也推荐大家读一读此文,使用Multiwfn有更加丰富灵活的绘制光谱的功能。
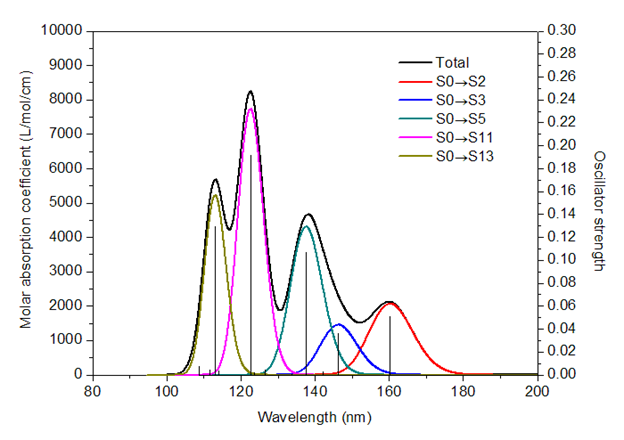
苯在乙醇中紫外-可见光谱的计算分两步进行:
(1)优化基态结构,输入文件为:
#p m062x/6-311G** opt scrf(smd,solvent=ethanol)Title Card Required
0 1
C -0.67502987 2.34767022 0.00000000
C 0.72013013 2.34767022 0.00000000
C 1.41766813 3.55542122 0.00000000
C 0.72001413 4.76393022 -0.00119900
C -0.67481087 4.76385222 -0.00167800
C -1.37241187 3.55564622 -0.00068200
H -1.22478887 1.39535322 0.00045000
H 1.26963813 1.39515722 0.00131500
H 2.51734813 3.55550122 0.00063400
H 1.27021413 5.71607322 -0.00125800
H -1.22493287 5.71613322 -0.00263100
H -2.47201587 3.55582922 -0.00086200
(2)取优化后的结构做激发态计算,输入文件为:
#p pbe1pbe/def2tzvp scrf=(smd,solvent=ethanol) td(nstates=20)Title Card Required
0 1
C 1.27035600 -0.57079400 0.00000100
C 1.12950000 0.81479100 -0.00002800
C -0.14082100 1.38551200 0.00002300
C -1.27038200 0.57073600 -0.00000500
C -1.12953700 -0.81473900 -0.00002000
C 0.14088500 -1.38550600 0.00002100
H 2.25940600 -1.01517400 0.00000400
H 2.00887500 1.44909000 -0.00000600
H -0.25069100 2.46420900 0.00003500
H -2.25937000 1.01525600 0.00001200
H -2.00882600 -1.44916100 -0.00002300
H 0.25060900 -2.46421700 0.00003100
注意:激发态的计算可以与几何结构优化在不同的水平。一般几何结构优化会用稍低的水平,而激发能的计算用较高的水平。由于紫外-可见光谱是众多吸收的加和,因此往往计算较多的态,本例计算了20个态。
TD-DFT的计算过程是先做基态计算,再基于基态的波函数进行激发态的计算。在激发态计算的输出文件中,最重要的部分如下:

使用GaussView打开输出文件,可以在Results→UV-Vis...部分查看光谱图,如下所示:
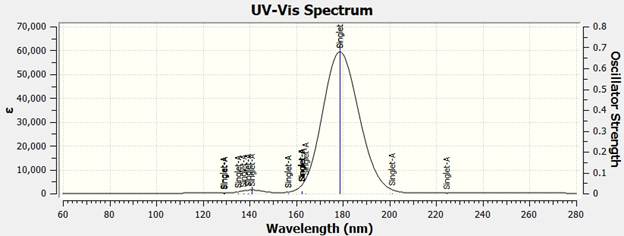
吸收峰的强度正比于振子强度,由于S1和S2的振子强度为0,因此最大吸收峰为S3对应的吸收(S4对应的吸收为178.68 nm,与S3近简并),位于179 nm处,与实验值185 nm非常吻合。
本文介绍了激发态计算的基本概念。近期我们还会继续推送一些激发态计算的教程,例如荧光的计算、磷光的计算、自然跃迁轨道分析等等。