在前几期的内容中,我和大家详细介绍了如何使用TwoSampleMR包读取暴露文件、去除存在连锁不平衡的SNP以及提取IV在结局中的信息,今天米老鼠将和大家介绍一下拿到数据后如何计算MR的结果并正确进行解读。
首先咱们运行一下之前的代码:
library(TwoSampleMR)
bmi_exp <- extract_instruments(
outcomes='ieu-a-835',
clump=TRUE, r2=0.01,
kb=5000,access_token= NULL
)
dim(bmi_exp)
# [1] 80 15
t2d_out <- extract_outcome_data(
snps=bmi_exp$SNP,
outcomes='ieu-a-26',
proxies = FALSE,
maf_threshold = 0.01,
access_token = NULL
)
dim(t2d_out)
# [1] 80 16现在我们准备好了暴露和结局的信息了,接下来要做一步很重要的工作,米老鼠理解为将IV的效应等位基因(effect allele)对齐,英文就是harmonise,具体使用的就是TwoSampleMR包的harmonise_data()函数,具体代码如下:
mydata <- harmonise_data(
exposure_dat=bmi_exp,
outcome_dat=t2d_out,
action= 2
)函数harmonise_data()只有三个参数,第一个和第二个参数分别是指定暴露数据和结局数据的,第三个参数action最重要,一般我们推荐使用默认值action=2即可,当然也可以使用action=3,这时候就表示去除所有存在回文结构的SNP。
注意了,mydata数据框有一列叫“mr_keep”,只有mr_keep是TRUE的SNP才是真正用于计算MR结果的IV。如果mr_keep是FALSE的话,那就说明这个SNP在计算MR结果时会被剔除。这个其实有点坑,会让很多刚入门的小伙伴认为mydata的所有SNP都是用来计算MR结果的。
最后,咱们只要简单使用mr()函数即可,代码如下:
res <- mr(mydata)
res具体的输出结果如下图所示:
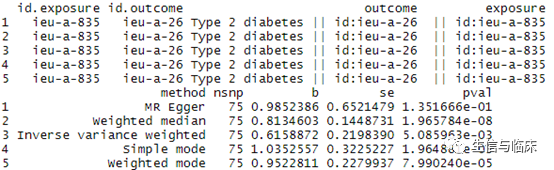
从图中我们不难看出,5种方法计算出来的结果是比较一致的,虽然MR Egger方法不显著,但是Inverse variance weighted (IVW)和Weighted median都是显著的,并且五个结果的效应值(b)都是正值,说明BMI的增加能导致糖尿病风险的升高。如果写到文章里的话,我们具体描述一下IVW的结果即可。
注意了,如果结局是一个二分类变量(通常是疾病),我们需要把b转化成OR,也即取b的自然指数即可。
除了上述5种计算方法外,TwoSampleMR包还提供了很多计算方法,比如随机效应模型和固定效应模型等,感兴趣的朋友可以通过mr_method_list()函数来了解。