今天给大家介绍由英国欣克斯顿,欧洲生物信息学研究所Ricard Argelaguet等人在《Nature Biotechnology》上发表了一篇名为“Computational principles and challenges in single-cell data integration”的综述。文中作者介绍了支持单细胞数据集成技术的基本概念,并讨论了用于链接不同数据集的锚的替代选择。此外,作者还回顾了单细胞数据集成策略的既定原则,局限性和诊断性,并强调了单细胞性状遗传分析方法和分子层间调控依赖性推断方法之间的相似性。最后,作者将基本的数据整合概念扩展到更具挑战性的未来应用,包括单细胞组学数据与物理维度(如空间和时间)的整合以及为个性化医疗构建人类变异参考图谱。

1
背景
多模式测序最有前途的特点之一是有机会从描述性的“快照”发展到对基因调控的机械理解。通过整合分子层之间的层次关系(即生物学的中心教条)的先验知识,多模式分析将在鉴定基因调控网络中事件的因果链方面发挥重要作用。
然而,单细胞多模态数据分析的一个关键挑战是设计有效的计算策略来整合不同的数据模式。数据集成通常被用于描述该任务的算法和软件,包括基于不同原理和假设的各种不同的计算策略。需要为这些数据集成任务定义统一的概念,以便根据输入数据结构和手头的特定集成任务将现有和未来的策略具体化。
2
输入数据和锚点的定义
任何数据集成管道中的第一步都是选择锚点以链接不同的数据模式。实际上,这种选择通常是由实验设计决定的,但它对下游分析具有根本影响,因为对锚的不同选择需要不同的统计和生物学假设,因此需要量身定制的方法。根据锚点的选择,可以区分三种类型的数据集成策略(图1):
基因组特征作为锚点(水平整合)
用于从独立的细胞组(不匹配的分析)分析相同数据模型的实验设计。比如:单细胞RNA测序(scRNA-seq)实验,通过不同的供体组中分析来自同一组织的细胞,或者通过结合不同scRNA-seq技术来组合数据,在这些实验中,分析是由它们的共同基因集锚点确定的。
细胞作为锚点(垂直整合)
用于同时从同一细胞分析多种数据模式的实验设计(匹配分析)。例如单细胞甲基组和转录组测序(scM&T-seq6),基因型、表达和甲基化的单细胞分析(sc-GEM16),通过测序(CITE-seq5)和单核染色质可及性和RNA表达测序(SNARE-seq17和SHARE-seq4)对转录组和表位进行细胞索引。其中,垂直整合的方法可以进一步分为局部和全局的方法。
高维空间无锚点(对角线整合)
用于不同实验间细胞和基因组特征不同的实验设计。例如,将scRNA-seq和单细胞转座酶可及染色质测序分析(scATAC-seq)应用于不同的细胞组。

图1 用于数据集成的锚的替代选择
3
数据集成存在一些统计挑战
(1)异构数据模式。使用不同分析方法收集的分子读数通常具有不同的统计特性,并且需要具有不同统计假设的定制方法。
(2)过度拟合。随着分子层数量的增加(以及特征的数量),建模策略面临着过度拟合的风险。
(3)缺失数据。与某些单细胞方法有关的一个主要问题是大量的信息丢失。重要的是,分析方法在定义丢失数据的方式方面有所不同。
(4)生物噪音和技术噪音。来自复杂实验设计的多组学数据集通常包含多种异质性来源,包括技术和生物学。
(5)可扩展性。随着测序成本的降低和技术的进步,我们预计多模式数据集将遵循与scRNA-seq相似的趋势,在不到10年的时间里,实验规模将从数千万个细胞增加到数百万个细胞。查询非常大的数据集需要快速的计算方法,这些方法通常依赖于随机推理模式。
(6)检测噪音。由于少量的起始材料,单电池技术固有地嘈杂并且导致大量的技术噪声。
(7) 原则上验证和评估模型输出。评估数据集成输出是最具挑战性的步骤之一。
4
分子测量与物理维度的集成
用于单细胞基因组学的大多数集成方法都没有明确考虑物理维度,例如时间和空间。然而,一些实验方案已经允许在单细胞水平上进行分子测量,同时保持有关细胞在感兴趣组织中的位置的一些信息,作者考虑在时间分辨和空间解析的多模式数据背景下数据集成的原则和挑战。
整合时间分辨数据
在对时程实验中的样本进行积分时,大多数水平积分方法都是独立地处理每个样本,而忽略了时间分量。如果从一个相似的时间点或一个稳定的状态对细胞群进行采样,这是一个合理的策略。然而,在某些情况下,考虑时间变化是必要的,特别是对于研究动态生物过程,如胚胎发育(图2)。
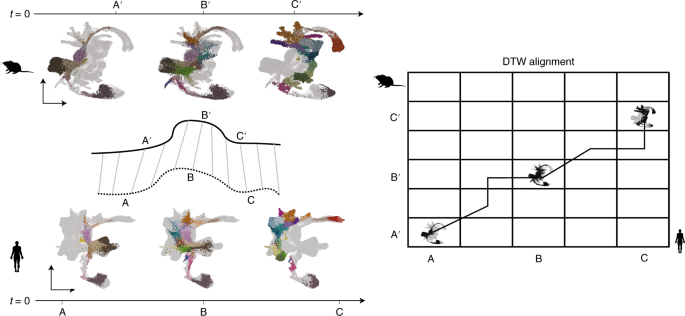
图2 绘制跨物种的时间分辨单细胞基因组学实验图
整合空间解析的单细胞数据
单细胞组学的应用有助于提高对细胞生物学的理解。然而,细胞从其固有的空间环境中分离,导致我们理解细胞通讯和器官功能的内在和外在因素之间相互作用的能力受到根本限制。由于在这两层之间缺乏简单的锚定选择,因此从组织病理学和显微镜分析中弥合单细胞分子读数和组织水平变化之间的差距一直具有挑战性。最近在多重成像和测序方面的技术进步允许在原位对单个细胞中的大量基因进行量化,并有望使这种多尺度建模成为现实。然而,从计算的角度来看,高分辨率分子信息和空间信息的整合带来了挑战,分析工具也刚刚开始出现。
5
个性化医学的多尺度集成
在人类健康背景下建立单细胞变异图谱的巨大努力将被证明是在细胞水平上理解疾病异质性的关键。然而,查询这些数据集将需要一套新的数据整合方法,将单细胞水平的变异与人类群体的生物医学特征联系起来。直观地说,这种多尺度方法的目标是从细胞表征中提取信息,解释人类个体水平上的表型变异。如果临床数据可以作为一个预测指标,这个任务可以被描述为一个有监督的学习问题,其中协变量对应于从细胞表征中提取的特征。这些特征可以使用基于模型的方法提取,也可以由专家知识手动定义。更普遍地说,我们设想,利用统计模型从单细胞表征中提取可解释和预测的特征将是一个活跃的研究领域。如果一个预测值只能作为一个多维变量使用,那么这个任务就变得更具挑战性,因为两个多维数据集必须绑定在一起。在这种情况下,两种表征之间的锚定是研究中的人类个体,因此定义了一种新型的整合问题,其中细胞和基因不能用作锚点(图3)。
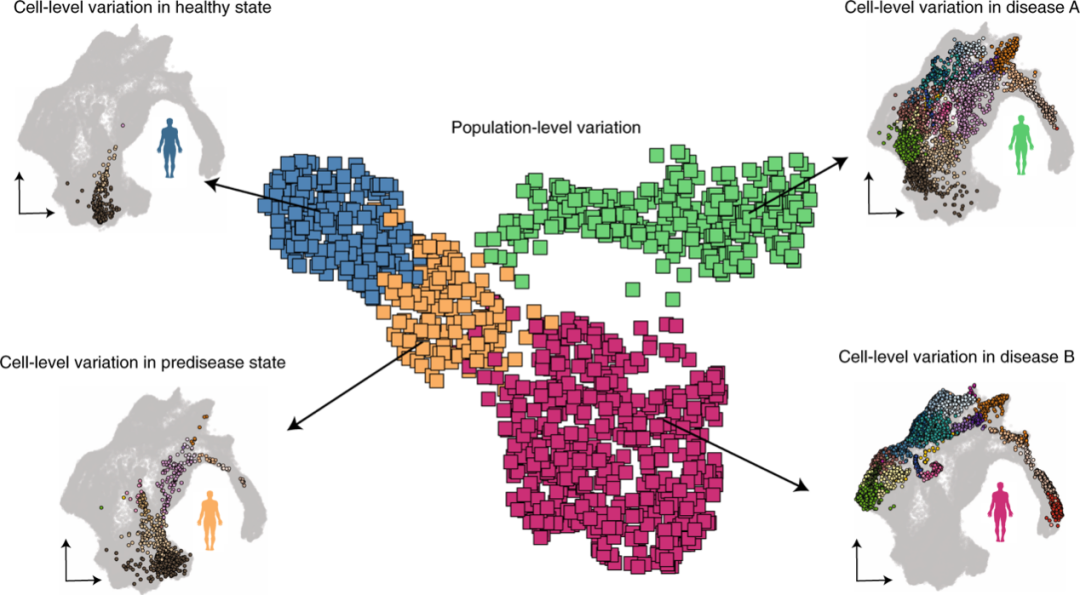
图3 利用单细胞分辨率的分子变异构建人类表型变异的种群水平图
6
总结
这篇综述中,作者介绍了一套单细胞数据集成技术的基本概念,并讨论了用于链接不同数据集的锚点的替代选择。作者回顾了数据集成策略的既定原则,局限性和诊断性,并着重介绍了单细胞性状遗传分析方法与分子层之间调节依赖性推论之间的相似之处。最后,作者将基本数据集成概念扩展到更具挑战性的未来应用中,包括将单细胞组学数据与物理维度集成在一起,以及为个性化医学构建人类变异参考图谱。
参考资料
Argelaguet, R., Cuomo, A.S.E., Stegle, O. et al. Computational principles and challenges in single-cell data integration. Nat Biotechnol (2021).
https://doi.org/10.1038/s41587-021-00895-7