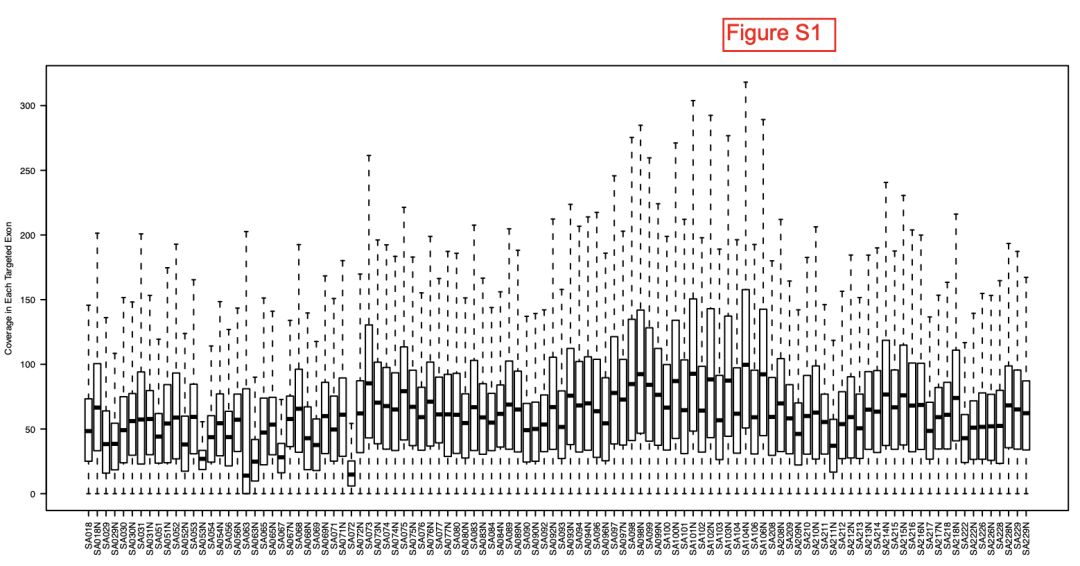
如果学徒之后跑流程,那其实前途很有限,所以我安排了一个随机任务,考核他们查资料解决问题的能力。我在Published: 04 April 2012 文章, The clonal and mutational evolution spectrum of primary triple-negative breast cancers 看到了一个有趣的图。
首先走wes流程拿到bam文件
这个我们多次讲解了,略,大家自行前往B站看WES视频:
然后根据CCDS数据库拿到人类全部exon的坐标在生信技能树早期教程我也多次讲解过,如何根据CCDS数据库文件,来制作如下BED格式的人类外显子坐标记录文件:
代码语言:javascript
复制
$ head hg38.exon.bed
chr1 69090 70007 OR4F5 0 +
chr1 450739 451677 OR4F29 0 +
chr1 685715 686653 OR4F16 0 +
chr1 801942 802433 LINC00115 0 +
chr1 925941 926012 SAMD11 0 +
chr1 930154 930335 SAMD11 0 +
chr1 931038 931088 SAMD11 0 +
chr1 935771 935895 SAMD11 0 +
chr1 939039 939128 SAMD11 0 +
chr1 939274 939459 SAMD11 0 +
使用samtools工具对exon坐标全部碱基计算覆盖深度
很简单的命令:
代码语言:javascript
复制
~/miniconda2/envs/WES/bin/samtools depth -b hg38.exon.bed a5.sort.bam > /tmp/tmp.depth
代码语言:javascript
复制
$ head tmp.depth
chr1 69091 5
chr1 69092 5
chr1 69093 5
chr1 69094 5
chr1 69095 4
chr1 69096 4
chr1 69097 4
chr1 69098 4
chr1 69099 4
chr1 69100 4
使用bedtools把碱基覆盖深度归属于exon
可以看到每个exon的所以坐标都是有测序深度的,这个文件目前是几千万行!
代码语言:javascript
复制
chr1 69090 70007 OR4F5 0 + chr1 69091 69091 5
chr1 69090 70007 OR4F5 0 + chr1 69092 69092 5
chr1 69090 70007 OR4F5 0 + chr1 69093 69093 5
chr1 69090 70007 OR4F5 0 + chr1 69094 69094 5
chr1 69090 70007 OR4F5 0 + chr1 69095 69095 4
chr1 69090 70007 OR4F5 0 + chr1 69096 69096 4
chr1 69090 70007 OR4F5 0 + chr1 69097 69097 4
chr1 69090 70007 OR4F5 0 + chr1 69098 69098 4
chr1 69090 70007 OR4F5 0 + chr1 69099 69099 4
chr1 69090 70007 OR4F5 0 + chr1 69100 69100 4
对exon进行汇总
每个坐标的测序深度取平均值即可,可以写一个简短的perl脚本,或者直接读入该文件到R语言,总之对20多万个外显子都计算一个平均测序深度即可。
绘制boxplot
这个是最简单了,参考文献里面的一百多个wes样本合并的boxplot。
课程内容 | |
|---|---|
1 | 生信-R语言入门 |
2 | GEO数据库挖掘 |
3 | 生信-LINUX基础 |
4 | 转录组课题设计和流程分析 |