题目:对配合物[Fe(H2O)6]2+在CASSCF(6,5)/def2-SVP水平计算能量最低的单态、三态、五重态能量。要求使用态平均的(state averaged) CASSCF,活性空间需要包含Fe的3d电子和3d轨道。 [Fe(H2O)6]2+结构选自文献J. Chem. Theory Comput.,16, 2224. (2020),见附录。
参考解答
使用BDF:
① 使用iCAS方法构造活性空间的初始猜测。BDF中的expandmo模块可以根据原子的AO基组自动确定 CASSCF活性空间和初始猜测轨道。详细介绍请参考
http://182.92.69.169:7226/expandmo
以及文献
J. Chem. Theory Comput., 17, 4846. (2021)
我们只需在输入文件中写出活性空间想要包含的AO基函数:
$compass title fe(h2o)62+ basis def2-svp geometry file=fe.xyz #附录中[Fe(H2O)6] 2+的xyz坐标文件 end geometry $end$xuanyuan
$end$scf
ROHF
charge
2
spin
5
molden
$end%cp BDF_WORKDIR/BDFTASK.scforb BDF_WORKDIR/BDFTASK.inporb
$expandmo
vcmo
minbas
5
1Fe|3D-2
1Fe|3D-1
1Fe|3D0
1Fe|3D1
1Fe|3D2
$end
输出文件给出的信息可用于MCSCF模块的输入文件。
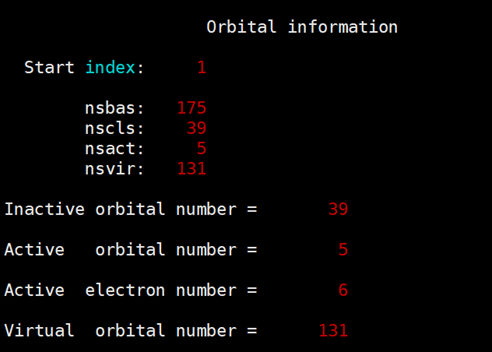
"nscls"后面列出的是非活性空间中每个不可约表示下的占据轨道数,"nsact"后面列出的是活性空间中每个不可约表示下的轨道数,这两行给出的信息可用于mcscf模块中"close"和"active"。计算产生的初始猜测轨道会保存在.exporb文件。使用关键molden后,会同时产生.exporb.molden文件,可用于检查exporb初始猜测轨道是否正确。</p><p>State averaged CASSCF计算的输入文件如下:</p><div class="rno-markdown-code"><div class="rno-markdown-code-toolbar"><div class="rno-markdown-code-toolbar-info"><div class="rno-markdown-code-toolbar-item is-type"><span class="is-m-hidden">代码语言:</span>javascript</div></div><div class="rno-markdown-code-toolbar-opt"><div class="rno-markdown-code-toolbar-copy"><i class="icon-copy"></i><span class="is-m-hidden">复制</span></div></div></div><div class="developer-code-block"><pre class="prism-token token line-numbers language-javascript"><code class="language-javascript" style="margin-left:0">compass
title
fe(h2o)62+
basis
def2-svp
geometry
file=fe.xyz
end geometry
saorb
$end
$xuanyuan
$end
%cp BDF_WORKDIR/BDFTASK.exporb BDF_WORKDIR/BDFTASK.inporb
$mcscf
guess
read #读取上一步计算的.exporb作为初猜
close
39
actel
6
active
5
mixci
3 #要算的根对应的不可约的种类数
1 3 5 #自旋多重度
1 1 1 #每个对应不可约和自旋多重度下要计算的根数
1 1 1 #要算的根分别对应的第几个不可约
molden
iprtmo
1
计算收敛后最好打开计算产生的.mcscf.molden文件,确认最后选进活性空间内的活性轨道。计算收敛后的轨道图如下:
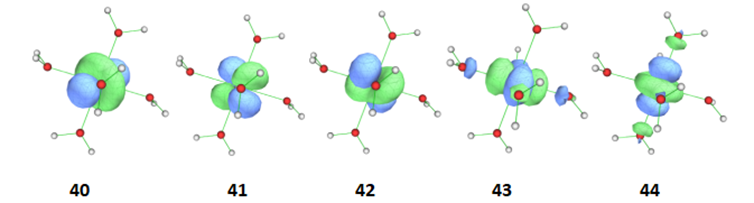
图1 [Fe(H2O)6]2+选进活性空间的轨道
最后得到的单重态、三重态和五重态的能量和组态信息如下:
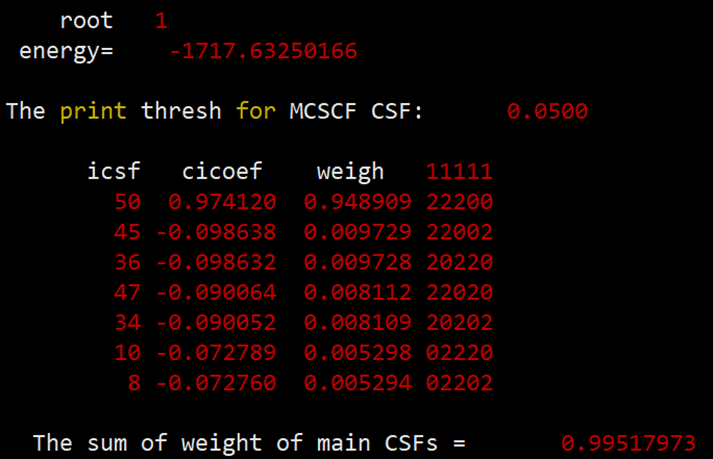
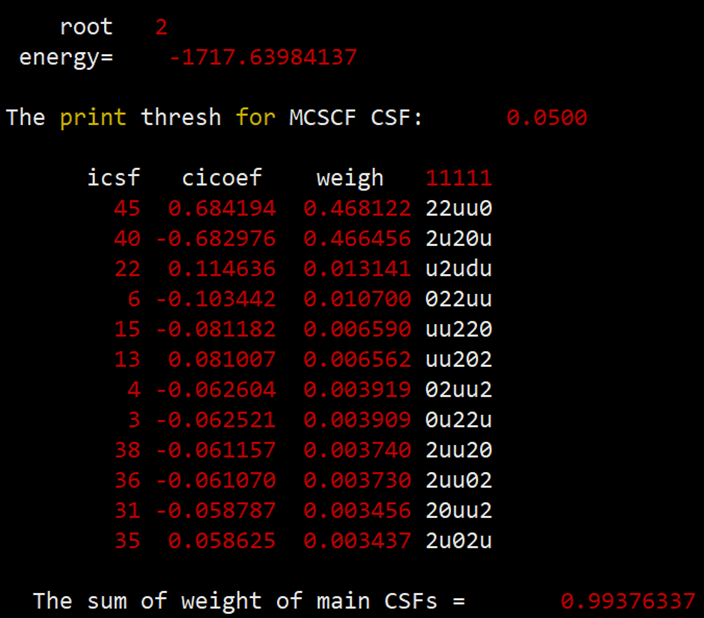
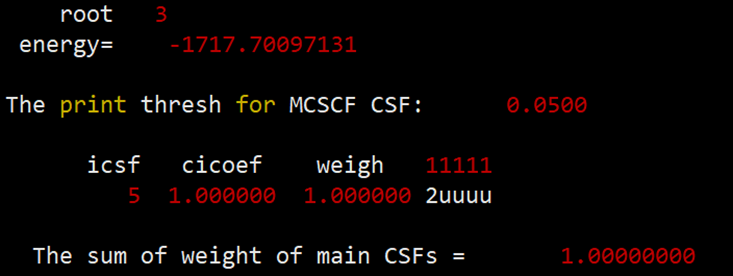
组态信息中的u代表的是自旋向上的单电子。
表1 使用BDF计算[Fe(H2O)6] 2+得到的能量(in Hartree)
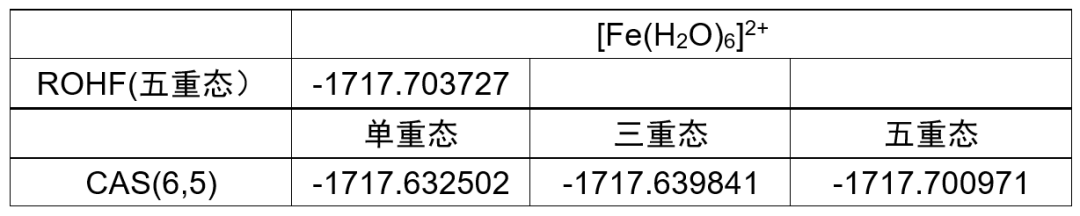
使用ORCA:
① 做ROHF计算,构造初始猜测轨道:
!ROHF def2-svp pal8
*xyzfile 2 5 fe.xyz #附录给的坐标
将计算产生的.gbw文件转换成molden,找出要选进活性空间的5条3d轨道及其编号。5条3d轨道及其编号如图2所示(轨道编号从1开始),因此不需要进行轨道交换。
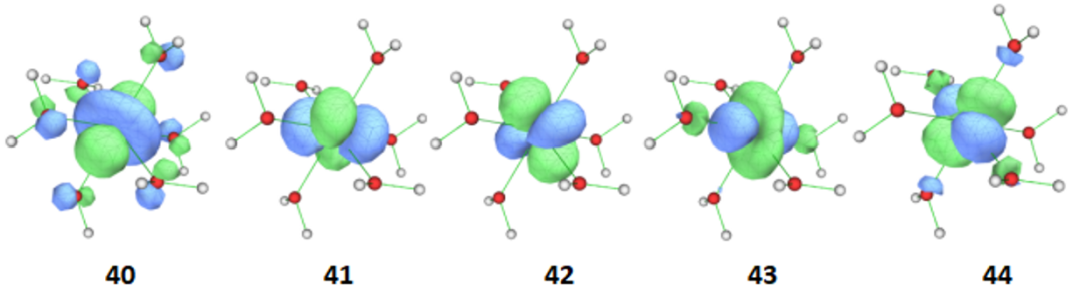
图2 [Fe(H2O)6]2+CASSCF计算想要选进活性空间的轨道
② State averaged CASSCF计算的输入文件为:
!def2-svp pal8
!moread
%moinp "rohf.gbw"
%casscf
nel 6
norb 5
mult 1,3,5
nroots 1,1,1
weights[0]=1 #该计算做的是state average CASSCF ,3个block的weight一样
weights[1]=1
weights[2]=1
end
*xyzfile 2 1 fe.xyz
在输出文件中可看出单重态、三重态和五重态各算了一个根,这三个根的权重相等,各占0.33333。
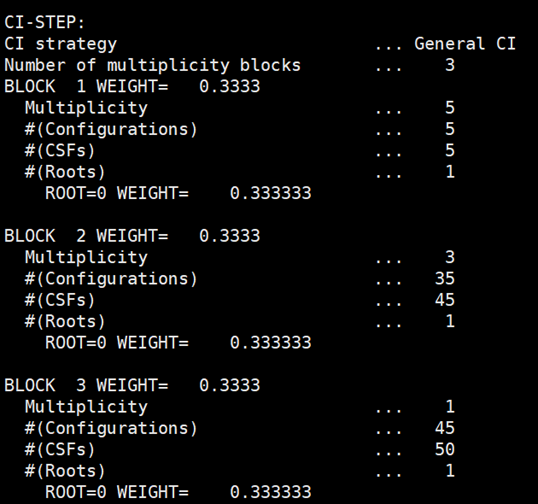
每个根的组态信息如下所示:
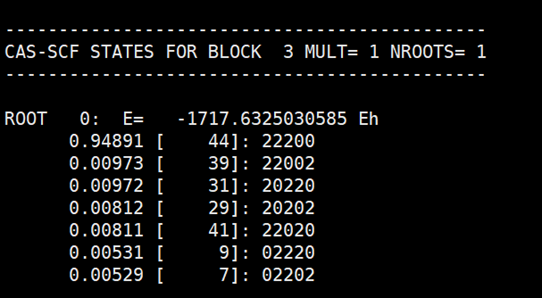
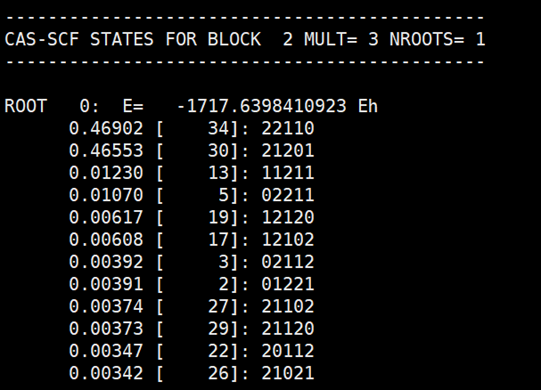

ORCA和BDF得到的结果完全一致。
附录
[Fe(H2O)6] 2+ 单位:Angstrom
Fe -0.64257176050830 0.51803920561668 0.11515259480577
O 0.11174687498777 2.15027362958097 -0.78003307659188
H 0.70766775417542 2.71590643817051 -0.25346860209865
O -1.31193844940493 -0.30254437211926 -1.59153974321848
H -1.93648366806763 0.13093135988795 -2.20090760771949
O 0.02720918798052 1.33940572113505 1.82122676893196
H 0.65258765622944 0.90686870076571 2.43040311553976
O -1.39648053530566 -1.11444807905756 1.01023916292248
H -1.99331392681300 -1.67940484230818 0.48397490889195
O -2.28682023617329 1.63340828371306 0.41005372492049
H -2.35377021104600 2.47291065207153 -0.08301808819513
O 1.00194372931180 -0.59712335622834 -0.17950165163229
H 1.06807766676477 -1.43758842942053 0.31202545851205
H -3.18922098471738 1.27280247171927 0.48036417156888
H -0.66521991563992 -0.78352006296416 -2.14179468111308
H 1.90454646424149 -0.23678289755293 -0.24845043594753
H 0.43776650602076 2.17113603799071 -1.69794346797289
H -0.61875604533523 1.82123954705219 2.37159866149671
H -1.72126010670064 -1.13595000805262 1.92856878689935