2022年3月23日,来自哥伦比亚大学的Artem Cherkasov和英伟达的Abraham C等人在Nature Machine Intelligence杂志发表文章,全面阐述了GPU计算和深度学习的历史趋势和最新进展,并讨论了它们对药物发现的直接影响。

摘要
深度学习几乎颠覆了每一个研究领域,包括那些对药物发现有直接重要性的领域,如药物化学和药理学。这场革命在很大程度上归功于高度可并行的GPU的空前进步和支持GPU的算法的发展。在这篇文章中,我们全面介绍了GPU算法的历史趋势和最新进展,并讨论了它们对发现新药和药物靶点的直接影响。我们还介绍了最先进的深度学习架构,这些架构在早期药物发现和随后的hit-to-lead阶段都有实际应用,包括加速分子对接、评估脱靶效应和预测药理特性。最后,我们讨论了GPU加速和深度学习模型对药物发现领域的全球民主化的影响,这可能会推动对不断扩大的化学世界的有效探索,以加速发现新药。
主要内容
图形处理单元 (graphics processing units, GPU) 最初是为加速三维图形而开发的,它在强大的并行计算方面的优势很快就被科学界所称赞。最早将GPU用于科学目的的尝试采用了可编程着色器语言来运行计算。2007年,英伟达公司发布了计算统一设备架构 (Compute Unified Device Architecture, CUDA) 作为C语言的扩展,同时还发布了编译器和调试器,为将计算密集型工作负载移植到GPU加速器中打开了闸门。进一步的进展来自于常见数学库的发布,如快速傅里叶变换和基本线性代数子程序,这些都是科学计算的基础。同年,第一批计算化学程序被移植到了GPU上,实现了分子力学和量子蒙特卡洛计算的高效并行化。
2014年9月,英伟达公司发布了cuDNN,这是一个由GPU加速的深度神经网络 (DNN) 基元库,实现了前向和后向卷积、池化、归一化和激活层等标准程序。GPU对训练和测试子过程的架构支持似乎对标准深度学习 (DL) 程序特别有效。因此,出现了一个由GPU加速的深度学习的整个生态系统。虽然英伟达的CUDA是一个更成熟的GPU编程框架,但AMD的ROCm代表了一个通用的GPU加速计算平台。ROCm引入了新的数值格式,以支持常见的开源机器学习库 (如TensorFlow和PyTorch),它还提供了将英伟达CUDA代码移植到AMD硬件的方法。值得注意的是,AMD不仅在GPU计算竞赛中追赶ROCm平台,而且最近还推出了新的旗舰GPU架构AMD Instinct MI200系列,与最新的NVIDIA Ampere A100 GPU架构竞争。
生物信息学、化学信息学和化学基因组学领域,包括计算机辅助药物发现 (CADD),已经利用了在GPU上运行的DL方法。CADD中的大多数挑战通常都面临着组合学和优化问题,而机器学习已经有效地提供了解决方案。因此,CADD应用中的DL已经取得了重大进展,如虚拟筛选、新药设计、吸收、分布、代谢、排泄和毒性 (ADMET) 特性预测等等 (图1)。
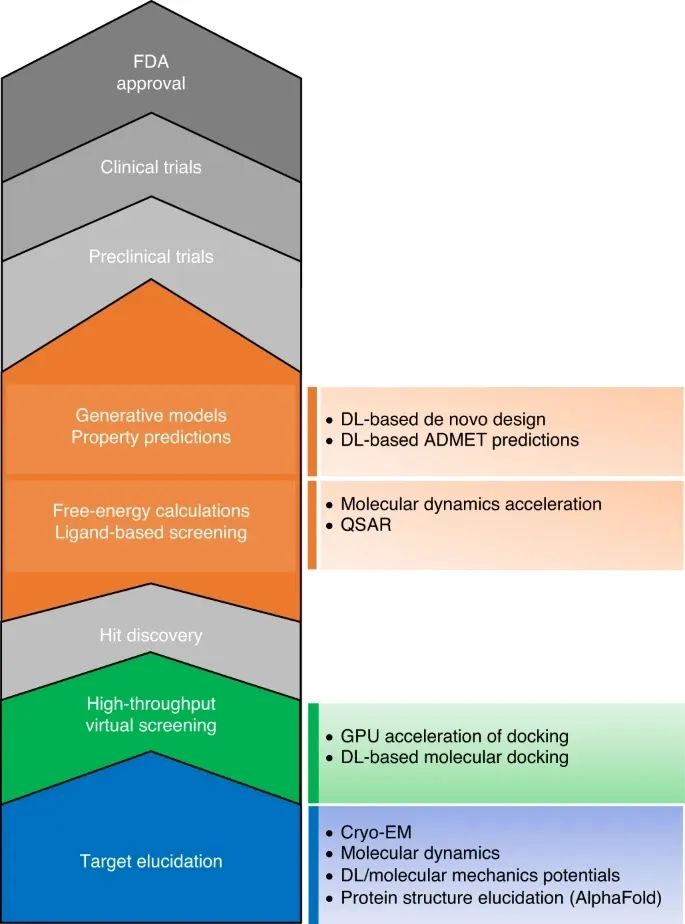
图1:CADD工作流程
GPU加速器在药物发现和开发过程的每个步骤中都能找到应用。
在此,我们讨论了GPU支持的并行化和DL模型开发和应用对蛋白质和蛋白质-配体复合物模拟的时间尺度和准确性的影响。我们还提供了用于低温电子显微镜 (cryo-EM) 结构测定和蛋白质三维结构预测中DL算法的例子。
用于分子模拟的GPU计算和深度学习
GPU的加速来自于大规模的数据并行性,它产生于对数据的许多元素执行的类似独立操作。在图形学中,一个常见的数据并行操作的例子是使用旋转矩阵跨越坐标,描述视图旋转时物体的位置。在分子模拟中,数据并行可以应用于原子势能的独立计算。同样,DL模型训练涉及到前向和后向的传递,这些传递通常表示为矩阵转换,是很容易并行化的 (图2)。
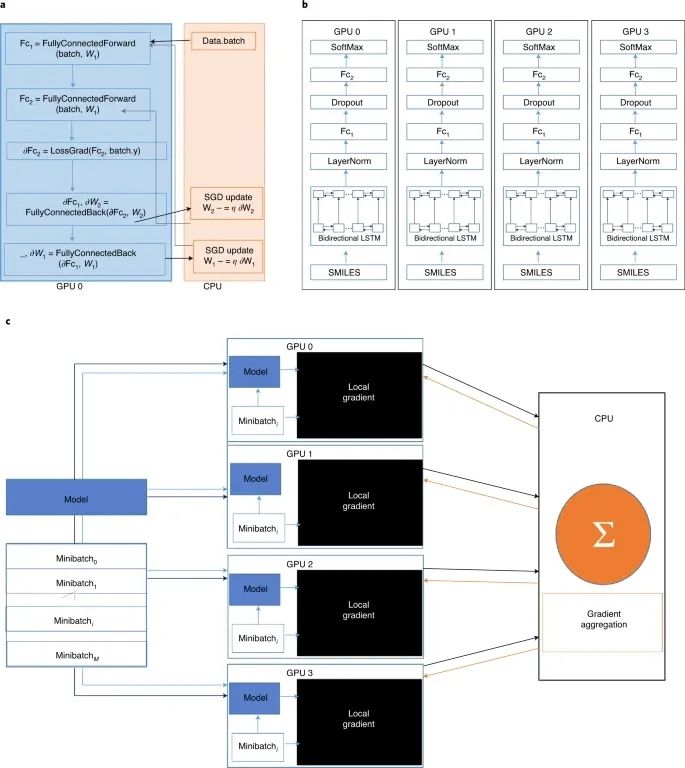
图2:单GPU和多GPU环境下DL架构的并行化
神经网络的算术运算是基于矩阵乘法,由GPU使用块乘法和聚合进行并行化。
a,两层多层感知器 (MLP) 的计算图在一个GPU上的分布。W,可训练参数;SGD,随机梯度下降算法;η,随机梯度下降算法的学习率。
b,数据并行化。每个GPU存储一个网络副本。数据并行化是最普遍采用的加速DL的GPU范式。网络的副本驻留在每个GPU中,每个GPU都有自己专用的小批数据来进行训练。然后将计算出的梯度和损失传输到共享设备 (通常是CPU) 进行聚合,然后再转播给GPU进行参数更新。LayerNorm、Dropout、Fc、SoftMax和Bidirectional LSTM (长短时记忆) 是用于演示的任意神经网络拓扑结构的模块。
c,梯度小批下降算法的正向和反向传播。M,数据的总小批量。
加速GPU上的分子动力学模拟
在过去的十年中,以GPU为中心的分子动力学代码的发展使得模拟的计算成本与基于中央处理器 (CPU) 的算法相比减少了数百倍。因此,大多数分子动力学引擎 (AMBER、GROMACS和NAMD) 现都提供GPU加速实现。GPU不仅非常适合加速分子动力学模拟,而且利用空间域分解,可以很好地扩展系统规模。因此,分子动力学模拟扩展到更广泛的生物分子现象,接近病毒和细胞水平,更接近于实验时间尺度。最近的方法和算法的进步使得分子动力学模拟的分子组合高达2×109个原子 (图3),总体模拟时间为微秒甚至毫秒。

图3:可以用分子动力学模拟的生物系统的复杂性的时间轴
多年来的持续开发努力,旨在用NAMD模拟复杂性不断增加的真实生物对象,从1990年代初期的千原子大小的小型溶剂化蛋白质到现在的十亿原子大小的完整原始细胞。ATP,三磷酸腺苷;HIV,人类免疫缺陷病毒;STMV,卫星烟草花叶病毒。
自由能模拟是另一个继续受益于GPU开发进展的领域。诸如相对结合自由能计算、热力学积分和自由能扰动等方法现在可以计算出大量蛋白质-配体复合物的可靠结合亲和力。在这方面,最近开发的基于神经网络的力场,如ANI和AIMNet提供了自由能模拟的工业标准精度。薛定谔 (Schrödinger) 基准集中的酪氨酸-蛋白激酶抑制剂的基准显示,使用ANI机器学习潜力的模拟将绝对结合自由能误差降低了50%。像ANI这样的框架为生成原子势提供了一个系统的方法,大大减少了拟合力场所需的人力,从而使力场开发自动化。最近,其他DL框架也被提出来,以进一步推动药物发现中的分子模拟的界限。作为这些方法的典范,加强采样的重加权自动编码器变异贝叶斯方法被成功地用于模拟配体-蛋白解离。它的处理速度明显快于传统的分子动力学,但却产生了准确的结合自由能和环形构象采样的估计。同样,Drew Bennett等人使用DNNs来预测来自分子动力学模拟的小分子的水-环己烷转移能量。在开源框架的支持下,也有人提出使用混合DL和分子力学势进行配体-蛋白质模拟。这些方法对配体采用基于量子力学的DL势,对周围环境采用分子力学势,与传统的势相比,在重现结合姿势方面显示出卓越的性能。
量子力学和GPU
CUDA和OpenCL应用编程接口 (API) 的出现是GPU应用成功的关键,尽管对GPU进行编程以高效运行化学代码并非易事。为了实现高效率,需要同时执行被分组为块的计算线程。TeraChem是第一个专门为GPU编写的量子化学代码。混合精度的算术允许非常有效地计算库仑和交换矩阵。TeraChem的最新算法发展允许用密度函数理论 (DFT) 模拟整个蛋白质。量子力学和分子力学的混合模拟,使人们对光激活机制有了深入的了解,并在分子水平上了解了光能转化为功的过程。DFT计算现在是研究蛋白质-配体相互作用的常规方法。例如,最好的计算结果是蛋白质-配体相互作用能量的平均绝对误差约为2 kcal mol-1。对丝氨酸蛋白酶X和酪氨酸蛋白激酶2的DFT计算表明,所得到的几何形状接近于共晶体的蛋白-配体结构。
未来的超大规模超级计算机将在异构的CPU和GPU环境中提供高水平的并行性。这种扩展需要开发新的混合算法,而且基本上是对科学代码的完全重写。这些新的发展现在正作为NWChemEx软件包的一部分来实施。NWChemEx将提供对系统进行量子力学和分子力学模拟的可能性,这些系统比理论方法的经典公式所能解决的问题大几个数量级。
蛋白质结构测定的GPU加速
冷冻电镜的高通量和自动化已经变得越来越重要,它是用于蛋白质结构测定的最先进的实验技术,可用于基于结构的药物设计。基于DL的方法,如DEFMap和DeepPicker,已经被开发出来以加速低温电镜图像的处理。DEFMap方法通过结合学习局部密度数据之间关系的DL和分子动力学模拟,直接提取与隐藏原子波动相关的结构动力学。DeepPicker采用卷积神经网络 (CNN) 和跨分子训练,从先前分析的显微照片中捕捉粒子的共同特征,这为单粒子分析中自动挑选粒子提供了便利。这个工具可以说明,DL集成可以成功地解决目前在实现全自动低温电镜管道方面的差距,为蛋白质科学的新的多学科方法铺平道路。
除了通过低温电镜加速蛋白质结构的实验表征外,最近DeepMind与AlphaFold-2方法在CASP挑战中取得的突破性成功,暗示了DL算法在蛋白质结构表征和可药用蛋白质组扩展中的未来影响。AlphaFold-2可以定期预测蛋白质的几何形状,并具有原子级的准确性,而无需之前接触过类似的结构。最近更新的基于神经网络的模型在大多数情况下表现出与实验相匹敌的准确性,并在第14届CASP竞赛中大大超过了其他方法。AlphaFold-2背后的DL模型结合了关于蛋白质结构的物理和生物知识,利用多序列比对来破解生物学中最古老的问题之一。AlphaFold-2被用来预测几乎所有已知的人类蛋白质和其他对医学研究很重要的生物体的结构,总共有35万个蛋白质,这对生物医学研究来说是一个了不起的成就。
CADD中DL的出现
DL的进展,特别是在计算机视觉和语言处理方面的进展,恢复了CADD研究人员最近对神经网络的兴趣。默克公司通过2012年的Kaggle分子活动挑战赛普及了CADD的DL。Dahl等人的获胜方案利用了多任务学习方法来训练DNN。此后,许多研究人员将这种模型用于药物发现问题。这些问题包括评估治疗药物的药代动力学行为及其不良反应的预测因素,预测小分子与蛋白质的结合,确定致癌细胞的化疗反应,药物敏感性的定量估计和定量结构-活性关系 (QSAR) 建模等等。
支持GPU的DL架构的出现,以及化学基因组学数据的激增,推动了有意义的CADD赋能的临床候选药物的发现。此外,人工智能驱动的公司 (如BenevolentAI、Insilico Medicine和Exscientia等) 正在报告AI促进药物发现方面的成功。例如,Exscientia开发了一种用于治疗强迫症的候选药物DSP-1181,从构思到使用人工智能方法不到12个月就进入了I期临床试验。Insilico Medicine公司刚开始用其第一个人工智能开发的候选药物进行临床试验,用于治疗特发性肺纤维化,BenevolentAI公司将baricitinib确定为COVID-19的潜在治疗药物。这些最近的成功案例表明,在GPU计算的支持下,进一步推广和应用人工智能驱动的方法可以大大加快发现新型和改进的药物。
用于CADD的DL架构
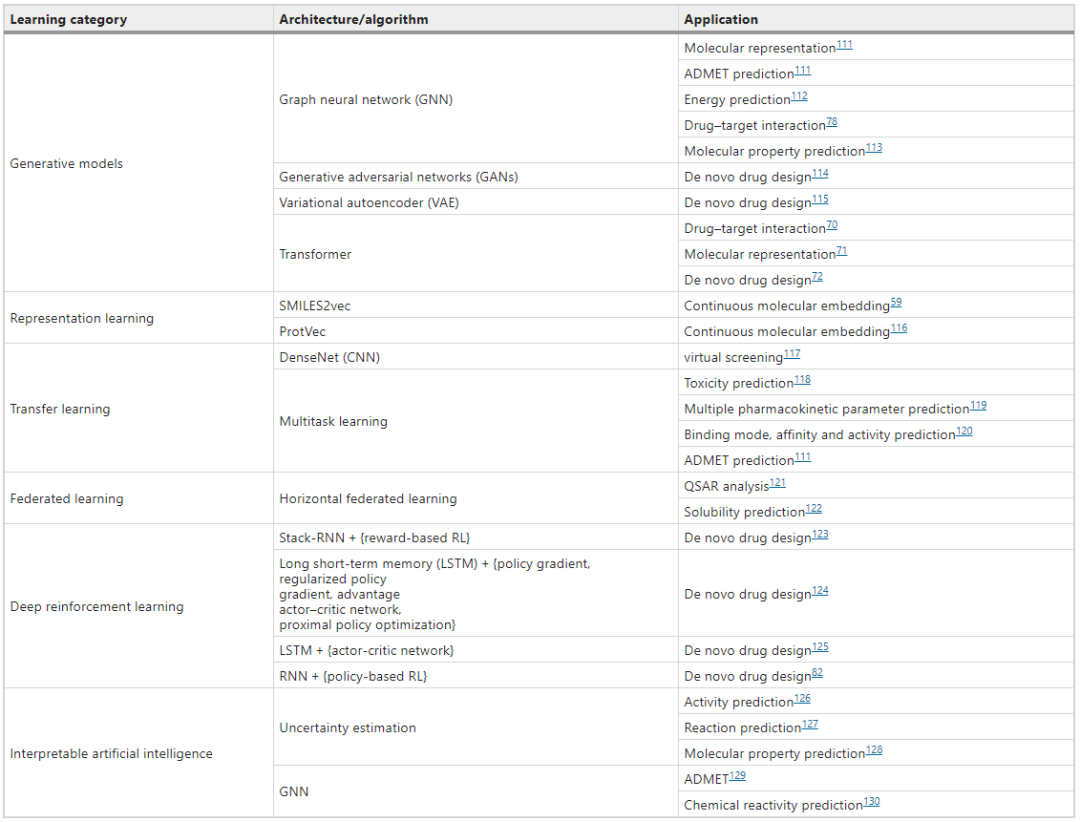
从应用于现有的或合成上可行的化学库的虚拟筛选的鉴别性神经网络,到最近DL生成模型的成功案例,激发了DL在新药设计中的应用。图4描述了常用的最先进的DL架构的一般方案。表1列举了它们在CADD中的应用。

图4:几种流行的神经网络的结构
a, Sigmoid神经元作为神经网络的构建块,是一个具有 sigmoid 非线性的感知器。
b, 一个全连接的前馈神经网络(MLP)由输入层、隐藏层和输出层组成,具有非线性激活功能 (如sigmoid)。X和Y分别代表模型的输入和输出。h,隐藏层;b,偏置项。
c, 一个简化的RNN的展开表示。U和W是可训练的模型参数;Si是RNN输入的"ith"时间步的潜在状态。
d,VAE。一个概率编码器在高斯假设下将输入映射到潜在空间。µ和∑是学习的多变量高斯分布的参数向量。从这个潜在空间中抽取样本,解码器试图从这些样本中重建原始输入。
e,CNN。核子对输入图像进行卷积,随后对特征图进行卷积,以逐步生成高阶特征图。池化进一步降低了特征图的维度。
f, GAN。鉴别器和发生器是两个任意的神经网络,它们在零和游戏中竞争,以合成新的样本。如果不使用硬件加速器 (如GPU),这些大容量的DL模型无法被合理地训练。这意味着 (除非另有说明) 这种模型被部署在GPU上。
表1 最先进的DL类别和它们在药物发现中的应用
MLPs
多层感知器 (Multilayer perceptrons,MLPs) 是具有输入、隐藏和输出层以及非线性激活函数 (sigmoid、tanh、ReLU等) 的全连接网络,是DNNs的基础。其较大的学习能力和相对较少的参数使MLPs成为人工神经网络在药物发现中最早的成功应用,用于QSAR研究。现代GPU机器使MLPs成为廉价的模型,适合于大型化学信息学数据集,对CADD产生了新的影响。
CNNs
CNNs可以说是使用最多的DNNs,它以分层原则为指导,利用小的感受野 (receptive fields) 来处理输入的局部子段。CNN一直是图像和视频处理的首选架构,同时它们也能在生物医学文本分类中取得成功。一个典型的CNN在三维体积 (高度、宽度、通道) 上运行,根据可学习的核子生成平移不变的特征图,并将这些图汇集起来以产生尺度和旋转不变的输出。
卷积操作的可并行性使CNN适合在GPU上实现。Toxic Color方法最初是利用Tox21基准数据开发的,使用的是简单的化合物二维图,证明了GPU支持的CNN预测,没有采用任何化学描述符,与最先进的机器学习方法相当。Goh等人随后介绍了Chemception,这是一个根据分子图训练的CNN,用于预测化学特性,如毒性、活性和溶解性,它显示出与用扩展连接性指纹训练的MLPs相当的性能。他们的模型通过将原子和键的特定化学信息编码到CNN中得到了进一步的改进。
RNNs
历史上,计算化学家广泛依赖拓扑学指纹,如扩展连接性指纹或其他描述符来描述分子特征。一种流行的线性Goh表示法是SMILES。固定长度的字符串表示很有用,因为它们可以被视为序列,并在时间网络中有效地建模,如循环神经网络 (recurrent neural networks,RNNs)。RNNs可以被看作是有记忆的马尔科夫链的延伸,能够通过其内部状态学习长距离的依赖关系,从而为分子序列的自动回归建模。
DL算法能够学习输入的分子的潜在内部表征,而不需要手工制作的描述符,这使得数据集和手头的问题在语法和语义上都有意义。SMILES2vec被训练从SMILES表征中学习连续嵌入,以对几个数据集和任务 (毒性、活性、溶解性和可溶性) 进行预测。这些向量的低维度加速了训练并降低了内存需求--这两者都是训练神经网络的关键方面。受流行的word-embedding算法word2vec的成功启发,Jaeger等人开发了mol2vec。基于在ZINC和ChEMBL数据集上对word2vec的无监督预训练,学到的表征达到了最先进的性能,并且比摩根指纹更适合于回归任务。
VAEs
变分自动编码器 (Variational autoencoders,VAEs) 是一种深度生成模型,由于其能够从观察到的数据中概率性地学习潜在空间,随后可以通过采样来生成具有微调功能特性的新分子,因此正在彻底改变化学信息学。VAEs支持直接采样,从而从潜在空间的学习分布中生成分子,而不需要昂贵的蒙特卡洛取样。Blaschke等人利用VAE模型生成了针对多巴胺受体2的新分子,这些分子被为活性预测而训练的支持向量机模型进一步验证。Sattarov等人探索了Seq2Seq VAEs来选择性地设计具有所需特性的化合物。一个生成的地形图 (topographic mapping) 被用来从VAE学到的潜在表征中采样。其他研究将VAE与分子图结合起来研究,以生成新分子。
GANs
最近,生成对抗网络 (generative adversarial networks,GANs) 已经确立了自己作为强大和多样化的深度生成模型的地位。GANs是基于生成器和鉴别器模块之间的对抗性游戏。鉴别器网络的目标是区分由生成器网络生成的真实和虚假数据点。一个同时训练的生成器网络试图创建新的数据点,从而使鉴别器被操纵,相信生成的结果是真实的。在GANs的经验性成功之后,人们提出了一些改进和修改意见。这些方法被药物发现的研究人员迅速利用,以人为地合成跨越子问题的数据。Méndez-Lucio等人在系统生物学和分子药物设计的交叉领域研究了一种基于GAN的生成模型方法。他们将生物学和化学结合起来的尝试体现在根据靶点的基因表达特征生成类似活性的分子。为此,他们使用了条件GANs和带有梯度惩罚的Wasserstein GAN的组合。GANs也已经与遗传算法结合起来进行探索,以防止模式崩溃,从而逐步探索更大的化学空间。
Transformer networks
在自然语言处理中使用Transformer networks所取得的巨大成功的启发下,药物发现领域的DL研究人员被激励着去探索它在训练序列的长期依赖性方面的能力。Shin等人利用自注意力机制(Self-Attention),进行了端到端的神经回归,以预测药物分子和靶点蛋白之间的亲和力分数。在此过程中,他们通过将分子标记嵌入与位置嵌入聚集在一起,为药物分子学习分子表征,并使用CNN为蛋白质学习新的表征。同样,Huang等人引入了MolTrans来预测药物与靶点的相互作用。Grechishnikova将特定靶点的分子生成制定为氨基酸链和它们的SMILES表示之间的翻译任务,并使用一个转换器编码器和解码器。
GNNs
最近在非欧几里得数据 (如图、点云和流形) 上使用DL的创新,促进了图神经网络 (graph neural networks,GNNs)。大多数GNN变体采取的核心形式是神经信息解析,其中来自图中每个节点的信息使用神经网络进行交换和迭代更新,从而产生稳健的表示。PyTorch Geometric通过利用稀疏的GPU加速为信息解析API提供CUDA内核。Deep Graph Library-LifeSci统一了几个开创性的工作,引入了一个平台无关的API,以便在生命科学中轻松整合GNN,特别是在药物发现方面。图形的数学表示法简洁地捕捉了分子的图形结构,这意味着GNNs在CADD中具有潜在的巨大用途。
Duvenaud等人的研究表明,在一些基准数据集上,药物的学习图表征优于圆形指纹 (circular fingerprints)。受门控GNN的启发,PotentialNet在基于配体的多任务 (电子特性、溶解度和毒性预测) 中表现出更好的性能。其他几项研究表明,当几何特征 (如原子距离) 也被考虑在内时,预测性能有所提高。Torng等人使用图自动编码器从氨基酸残基中学习蛋白质表征,以及蛋白质口袋的图表征。然后将这些向量与药物分子的图形表示相连接,并将其输入MLP,以预测药物与蛋白质的关系。Gao等人使用RNNs和GNNs分别对蛋白质序列和药物的原子图学习蛋白质和药物嵌入。一种报道的流行的药物再利用方法涉及知识图谱,这些大型知识图谱是由疾病、药物和适应症之间的已知相似性建立的。Gaudelet等人对GNN的CADD应用进行了广泛的回顾。
Reinforcement learning
强化学习 (Reinforcement learning) 是人工智能的一个分支,通过优化基于奖励和惩罚的策略来模拟决策。随着DL的渗透,深度强化学习已经在CADD中找到了应用,特别是在新药设计中,通过使分子具有理想的化学特性。在GNN上训练的深度强化学习被进一步证明可以提高生成的分子结构的有效性。强制执行有化学意义的行动,同时围绕化学性质优化奖励,产生有用的线索,将化学领域的知识传授给其他主要是黑箱的DL解决方案。
利用GPU和DL扩大虚拟筛选的规模
基于结构的虚拟筛选和基于配体的虚拟筛选旨在根据计算出的化合物与靶点的结合亲和力对其进行排序,并分别将小分子之间的结构相似性推断为功能等同性。随着可购买的配体库的指数式增长,已经包括数百亿的可合成分子,人们对扩大传统虚拟筛选的操作规模,对对接计算的并行化或基于DL的加速越来越感兴趣。
最近开发了一些基于结构的虚拟筛选方法,以有效地筛选数十亿条的化学库。VirtualFlow代表了这种平台的第一个例子,它允许在几个星期内在大型CPU集群 (约10,000个核心) 上筛选10亿个分子,同时显示出线性扩展行为。与VirtualFlow和其他基于CPU的方法不同,使用OpenCL和CUDA库对对接算法进行GPU加速,通过将整个蛋白质表面划分为任意独立的区域 (或斑点) 或在异构计算系统中结合多核CPU架构和GPU加速器,部分解决了高通量瓶颈问题。这种策略的一个最新例子是Autodock-GPU,它通过并行化姿势搜索过程,在大型GPU集群[如Summit超级计算机 (约27,000个GPU) ]上一天内可以筛选出10亿个分子。因此,这些在高性能计算上利用GPU计算的方法将可能成为从大型、多样的化学库中识别新的先导化合物,或加速其他基于结构的方法,如反向对接。然而,计算成本仍然很高,对于无法访问超级计算集群的药物发现机构来说,可能是难以承受的。
另一方面,最近出现了其他基于结构的虚拟筛选平台,利用DL预测和分子对接来促进从计算资源有限的大型库中选择活性化合物。这些方法的共同策略是实施经典计算筛选分数的DL仿真器,其推断速度比传统对接高一个数量级。预测性DL模型是使用各种化学结构表征建立的,从分子指纹到更复杂的嵌入,以过滤掉化学库的大部分分子。最早开发的方法之一,深度对接,依赖于一个完全连接的MLP模型,该模型用化学指纹和库中一小部分的分数来训练,然后用来预测剩余分子的对接分数等级,允许在不对接的情况下删除低排名条目。深度对接最初由Ton等人部署,使用Glide对SARS-CoV-2主要蛋白酶从ZINC15中筛选出13亿个分子。最近,Gentile等人还将其连续应用于不同的对接程序,以筛选出400亿个针对SARS-CoV-2主蛋白酶的市售分子,从而发现了新的经实验证实的抑制剂骨架。其他类似的方法也被提出来,这些方法依赖于预测对接结果的DL模型,如MolPAL和AutoQSAR/DeepChem。Hofmarcher等人也在ZINC数据库上进行了基于配体的虚拟筛选,该数据库有超过10亿个化合物,使用RNN对潜在的SARS-CoV-2抑制剂进行排序。与粗暴的方法相比,这些基于DL的方法可能在使学术研究小组和小型/中型企业都能获得化学空间方面发挥重要作用。
支持GPU的DL促进开放科学和药物发现的民主化
这里介绍的CADD中DL的集成,极大地促进了全球药物发现的民主化和开放科学的努力。开源的DL软件包DeepChem、ATOM、Deep Docking、MolPAL、OpenChem、GraphInvent和MOSES等,使用流行的机器学习库,包括 (但不限于) scikit-learn、Tensorflow和Pytorch,简化了DL策略与药物发现管线的整合。对DL模型的大数据集的需求不断增长,自然会鼓励数据共享的做法,并呼吁更广泛的开放数据政策。此外,云原生计算中的GPU加速和面向微服务的架构可以使CADD方法免费和广泛使用,有助于实现计算模块和工具,以及架构、平台和用户界面的标准化。DL解决方案可以利用公共云服务的优势,如亚马逊网络服务、谷歌云平台和微软Azure,通过降低成本促进药物发现。
尽管这些新的DL支持的建模机会令人兴奋,但CADD科学家需要对DL技术的预期影响持谨慎态度。现实的期望需要从20多年来数据驱动的分子建模的经验教训和最佳实践中得出。例如,数据的质量、数量和多样性不仅会妨碍CADD模型的准确性,也会妨碍其整体的通用性。因此,数据清理和整理将继续发挥重要作用,它可以直接决定这种DL应用的成功或失败。
另一方面,使用来自指导性实验或高水平计算机模拟的动态数据集可以促进主动学习策略的使用。交互式训练和验证可以大大提高模型的质量,正如AutoQSAR工具所实现的那样。除了预测模型,DL解决方案在结合生成模型和基于RL的决策方法时特别有用。对基于奖励和惩罚的规则进行优化,可以使具有所需化学和功能特性的化学结构得到前所未有的"点菜式"设计。这种在新药设计中同时强制执行化学和生物意义上的行动的方法,代表了与更传统的黑箱DL解决方案的巨大差异。
开放科学的努力正受益于最近的端到端DL模型,这些模型可以在药物发现的所有阶段使用GPU来实现。最近开发的一个这样的平台是IMPECABLE,它集成了多种CADD方法。Al Saadi等人将分子动力学在预测结合自由能方面的优势与对接在姿势预测方面的优势相结合。他们的解决方案不仅实现了虚拟筛选的自动化,而且还实现了lead的细化和优化。
NVIDIA Clara Discovery是一个由GPU加速的框架、工具和应用程序组成的集合,用于计算药物发现,涵盖分子模拟、虚拟筛选、量子化学、基因组学、显微镜和自然语言处理。这些平台旨在开放和交叉兼容,并有望加速整个生物制药领域不同数据源的整合,从研究论文、病人记录、症状和生物医学图像到基因、蛋白质和候选药物。
许多主要的硬件生产商现在利用他们的计算专长,通过采用多个GPU集群来训练大容量的DL模型,用于反应预测、分子优化和新分子生成,从而进入超级计算的领域。CADD平台对制药终端的DL模拟的采用,可以使包含数百亿化合物的药库上的药物发现变得可行,即使是那些没有获得精英计算设施的小公司和学术实验室。
由于法律上的复杂性,机构间共享专有数据仍然是简化药物发现研究的瓶颈。联邦学习允许参与机构在各自的非共享数据上进行本地化训练。训练好的本地模型然后聚集在一个中央服务器上,以便更广泛地访问。因此,联邦学习通过在一定程度上缓解数据交换的挑战来支持民主化,尽管有效的模型聚合仍然是一个活跃的研究领域。
结论和展望
现代药物发现已经受益于最近DL模型和GPU并行计算的爆炸性增长。在硬件进步的推动下,DL在药物发现问题上表现得非常出色,从虚拟筛选和QSAR分析到生成性药物设计。特别是新药设计一直是GPU计算进步的主要受益者之一,因为它利用了大容量和高参数化的模型 (如VAE和GANs),不使用GPU等硬件加速器是无法合理部署的。近年来,GPU硬件的性价比不断提高,DL对GPU的依赖,以及DL在CADD中的广泛采用,都体现在CAS中超过50%的"化学中的人工智能"文献是在过去4年中发表的。此外,混合的人工智能方法已经被采用,它将传统的分子模拟与DL相结合,用于快速准确地筛选接近数千亿分子的超大型化学库。我们预计,越来越强大的GPU架构的可用性,加上先进的DL策略和GPU加速算法的发展,将有助于使全世界更广泛的科学界能够负担得起和获得药物发现。
DL算法的另一个关键驱动力是"大数据"的可用性。随着基因测序和高通量筛选的日益便捷,大量的原始数据现在很容易被数据驱动的计算化学的研究人员获得。然而,对于监督学习方法来说,高质量的标记数据仍然是昂贵的。因此,建立在从辅助数据集中学习的方法、使用迁移学习的知识转移和零样本学习等标签保守方法成为药物发现的DL的核心部分。为药物发现而开发的任何DL方法的可靠性和可推广性,关键取决于来源数据的质量。因此,数据清洗和整理发挥着重要作用,可以完全确定这种DL应用的成败,因此,深入探索集中的、经过处理的和良好标记的数据库的所谓好处仍然是一个开放的研究领域。
总的来说,药物发现和机器学习的研究人员已经有效地合作,以确定CADD的子问题和相应的DL工具。我们相信,在未来的几年里,这些应用将得到微调和成熟,这种合作将进一步发展到生命科学的其他未探索的领域。因此,联邦学习和协作式机器学习正在获得牵引力,我们相信它们将成为民主化药物发现革命的先声。
参考资料
Pandey, M., Fernandez, M., Gentile, F. et al. The transformational role of GPU computing and deep learning in drug discovery. Nat Mach Intell 4, 211–221 (2022). https://doi.org/10.1038/s42256-022-00463-x
--------- End ---------