背景
一、基因预测
在得到了一个物种的基因组序列之后,就可以开始对其序列进行分析了。序列分析主要包括结构基因组分析,功能基因组分析以及比较基因组分析几部分。通过对序列进行全面地分析,在基因组水平上了解一个物种的特点。序列分析主要包括基因预测,基因功能注释,ncRNA,重复序列,特殊功能序列,比较基因组等方面。
二、如何学习生物软件?
生物信息主要都是通过软件完成的,软件是集成了数据处理的算法规则。前面的内容中那个已经学习了大量生物软件的使用,本节内容将系统的总结一下生物软件的使用。
学习和使用一款生物软件主要需要遵守以下规则:
1、首先,要清楚自己的分析目的,然后选择合适的软件,一般文件资料选择最佳工具,比如原核基因预测可以选择使用 prodigal 软件;
2、一般分析最好同时使用多个软件,方便比较结果,例如原核基因预测可以使用 prodigal,glimmer3 以及 genemark 等,之前 NCBI 给的结果包括这三个软件的结果;
3、选择软件之后要能找到软件的官方网站:例如prodigal:https://www.psc.edu/resources/software/prodigal/
4、通过网站找到软件的介绍信息,安装说明,帮助文档,安装软件包;
5、找到软件发表文章;
6、正确安装软件,并能找到案例数据以及版本信息;
7、通过 bioconda 查找并安装软件;
8、知道软件的输入文件和输出文件以及使用范围;
9、找到软件的选项参数,并运行软件;
10、能够读懂软件输出结果;
三、原核生物基因预测
3.1 开放阅读框
开放阅读框指的是从 5'端开始翻译起始密码子(ATG)到终止密码子(TTA、TAG、TGA)的蛋白质编码碱基序列。每个序列都有 6 个可能的开放阅读框,其中 3 个开始于第 1、2、3个碱基位点并沿着给定序列的 5'→ 3'的方向进行延伸,而另外的 3 个开始于第 1、2、3 个碱基位点但沿着互补序列的 5'→ 3'的方向进行延伸。在开始这项工作之前,我们并不知道DNA 双链中哪一条单链是编码链,也不知道准确的翻译起始点在何处,由于每条链都有 3种可能的开放阅读框,2 条链共计 6 种可能的开放读框,我们的目的就是从这 6 个可能的开放阅读框中找出一个正确的开放阅读框。根据这个开放阅读框翻译得到的氨基酸序列才是真正表达的蛋白质产物。也就是软件会首先在序列中找开放阅读框 orf,开放阅读框 orf 可能是基因,也可能不是,理论上只有 1/6 的开放阅读框是基因。然后就是训练集,原核可以以自身序列作为训练集,真核比较复杂一些。训练集可以理解为软件需要先对其基因基本特征有所了解。
3.2 原核生物基因预测原理
原核生物一个完整的原核基因结构是从基因的 5'端启动子区域开始,到 3'端终止区域结束。基因的转录开始位置由转录起始位点确定,转录过程直至遇到转录终止位点结束,转录的内容包括 5'端非翻译区、开放阅读框及 3'端非翻译区。基因翻译的准确起止位置由起始密码子和终止密码子决定,翻译的对象即为介于这两者之间的开放阅读框 ORF。

原核生物 orf 结构
原核生物基因结构一般比较简单,基因是连续的,并不存在内含子。因此,在预测过程中相对于真核生物来说,相对容易一些。
3.3 密码子表
预测基因,需要注意的是要根据具体的物种选择合适的密码子表。
http://www.ncbi.nlm.nih.gov/Taxonomy/Utils/wprintgc.cgi
其中第一套是标准密码子表,第 11 套是细菌,古细菌以及植物线粒体。这里面需要注意,如果是支原体,一定要选择第四套密码子,因为它的起始密码子并不是 AUG,否则就会出现问题。
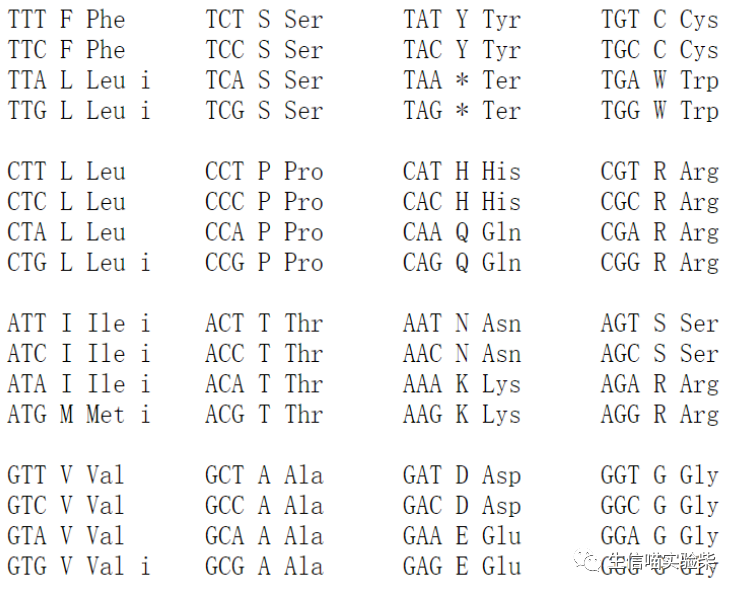
第四套密码子表
原核生物直接使用自身基因组作为训练集就可以,将基因组序列输入给软件,很快就会得到基因集的序列,这个分析并不困难。
原核生物的基因预测比较简单,准确性高,常用的软件包括 glimmer3,prodigal,genemark等工具。
四、利用 prodigal 预测基因
prodigal 的全称是 Prokaryotic Dynamic Programming Genefinding Algorithm,原核的动态编程基因查找算法,prodigal 主要应用于细菌和古生菌的基因预测,不能用于真核生物,如果要对 meta 样品做基因预测,prodigal 还专门提供了 meta 的版本。除此之外,prodigal还支持在线提交序列的方式来预测基因预测。也非常地易于使用。而且相对于 glimmer 基因预测工具,prodigal 更加好用,只需一步即可,而且,软件可以直接输出基因的核酸序列并翻译出的相应的氨基酸序列,这对很多初学者来说是非常方便的。
安装软件:
#软件安装
mamba install -y prodigal使用案例:
#1 prodigal基因预测
prodigal -a MGH78578.pep -d MGH78578.fnn -f gff -o MGH78578.gff -i MGH78578.fasta -c选项参数:
-a 是输出氨基酸文件-c 不允许基因一边断开,也就是要求完整的 orf,有起始和终止结构
-d 输出预测基因的序列文件
-f 选择输出文件格式,有 gbk,gff,和 sco 格式可供选择
-g 指定密码子,原核为第 11 套 -i 输入文件,即需要预测的基因组序列文件
-m 屏蔽基因组中的 N 碱基
-o 输出文件,默认为屏幕输出
-p 选择方式,是单菌还是 meta 样品
-q 不输错错误信息到屏幕
-t 指定训练集
-s 输出所有潜在基因以及分值到一个文件中
五、利用 glimmer3 预测基因
Glimmer 是用于原核生物基因组预测的工具,只要输入原核生物基因组即可得到其基因信息。不过该软件最终结果只是基因的位置信息,需要额外程序将基因从基因组上提取出来,并翻译成对应的氨基酸序列。
官方网站:http://ccb.jhu.edu/software/glimmer/index.shtml
glimmer 并不能像 prodigal 那样,一步完成工作。Glimmer 做基因预测一般需要 2 个步骤。首先是建立预测的模型,第二步是利用模型来对基因组进行基因预测。模型也叫训练集,也就是先让软件了解基因的一些特征,这样软件就能根据已知的信息,来推测未知的信息。
建立模型采用 build-icm 程序来完成。build-icm 的输入有三种。
1、某基因组的已知信息;
2、通过 long-orfs 产生的长的无重叠的 orfs;
3、高度相似的物种的基因。
可以选用自身作为训练集来作为模型。
软件安装:
mamba install -y glimmer软件使用:
#运行软件
sed -e '/>/d' MGH78578.fasta |tr -d '\n' |awk 'BEGIN {print ">wholefile"}{print $0}' >wholefile
long-orfs -n -t 1.15 wholefile tagname.longorfs 1>/dev/null 2>/dev/null
extract -t wholefile tagname.longorfs > tagname.train 2>/dev/null
build-icm -r tagname.icm < tagname.train 1>/dev/null 2>/dev/null
glimmer3 -o50 -g110 -t30 MGH78578.fasta tagname.icm refref.predict列表是结果。
六、在线预测
如果是少量序列,可以选择在线预测的方法。
原核基因预测:prodigal
网站链接:http://prodigal.ornl.gov/
原核基因预测:glimmer3
网站链接:http://ccb.jhu.edu/software/glimmer/index.shtml
国家微生物科学数据中心:prodigal
https://nmdc.cn/analyze/details?id=600671720b38496ee0c908dc
国家微生物科学数据中心:glimmer3
https://nmdc.cn/analyze/details?id=600671720b38496ee0c908dd
genemarks:http://exon.gatech.edu/GeneMark/gm.cgi
七、gff 与 gtf 格式文件
gff,gtf 与 bed 都属于基因组区间坐标文件,是生物信息分析中常用的三种列表格式。基因预测的结果,一般以 gff 格式展示。
7.1 GFF 格式
general feature format,是由 sanger 研究所定义,是一种简单的、方便的对于 DNA、RNA以及蛋白质序列的特征进行描述的一种数据格式,已经成为序列注释的通用格式,许多软件都支持输入或者输出 gff 格式。
基因组上每个功能区域成为一个 feature。gff 文件是一种用来描述基因组特征的文件,现在更新到第三版,通常称为 gff3。GFF 是文本文件,由 TAB 键隔开的 9 列组成,主要是用来存储基因组注释信息,可以直接使用 Excel 打开查看。
说明文档:https://github.com/The-Sequence-Ontology/Specifications/blob/master/gff3.md
网址:https://genome.ucsc.edu/FAQ/FAQformat.html#format1
seq_id:序列的编号,一般为 chr 或者 scanfold 编号;
source: 注释的来源,一般为数据库或者注释的机构,如果未知,则用点“.”代替
type: 注释信息的类型,比如 Gene、cDNA、mRNA、CDS 等;
start: 该基因或转录本在参考序列上的起始位置;(从 1 开始,包含);
end: 该基因或转录本在参考序列上的终止位置;(从 1 开始,包含);
score: 得分,数字,是注释信息可能性的说明,可以是序列相似性比对时的 E-values 值或者基
因预测时的 P-values 值,.表示为空;
strand: 该基因或转录本位于参考序列的正链(+)或负链(-)上;
phase: 仅对注释类型为“CDS”有效,表示起始编码的位置,有效值为 0、12. (对于编码蛋白质的
CDS 来说,本列指定下一个密码子开始的位置。每 3 个核苷酸翻译一个氨基酸,从 0 开始,CDS 的
起始位置,除以 3,余数就是这个值,,表示到达下一个密码子需要跳过的碱基个数。该编码区第
一个密码子的位置,取值 0,1,2。0 表示该编码框的第一个密码子第一个碱基位于其 5’末端;1 表
示该编码框的第一个密码子的第一个碱基位于该编码区外;2 表示该编码框的第一个密码子的第一、
二个碱基位于该编码区外;如果 Feature 为 CDS 时,必须指明具体值。);
attributes: 一个包含众多属性的列表,格式为“标签=值”(tag=value),以多个键值对组成的
注释信息描述,键与值之间用“=”,不同的键值用“;”隔开,一个键可以有多个值,不同值用“,”
分割。注意如果描述中包括 tab 键以及“,= ;”,要用 URL 转义规则进行转义,如 tab 键用 代替。
键是区分大小写的,以大写字母开头的键是预先定义好的,在后面可能被其他注释信息所调用。7.2 GTF 格式
GTF(GeneTransfer Format)格式与 GFF 格式类似,也是由 TAB 键分开的列表格式,一共由9 列组成,前 8 列与 GFF 格式相同,只是第九列不同,主要是用来对基因进行注释。其中的T 代表着转录本,也即是 GTF 中会列出一个基因的转录本组成。gtf 在 RNAseq 分析中要使用到。
1) seq_id:序列的编号,一般为 chr 或者 scanfold 编号;
2) source: 注释的来源,一般为数据库或者注释的机构,如果未知,则用点“.”代替;
3) type: 注释信息的类型,比如 Gene、cDNA、mRNA、CDS 等
4) start:该基因或转录本在参考序列上的起始位置;
5) end: 该基因或转录本在参考序列上的终止位置;
6) score: 得分,数字,是注释信息可能性的说明,可以是序列相似性比对时的 E-values 值或
者基因预测时的 P-values 值,“.”表示为空;
7) strand: 该基因或转录本位于参考序列的正链(+)或负链(-)上;
8) phase: 仅对注释类型为“CDS”有效,表示起始编码的位置,有效值为 0、1、2(对于编码蛋白
质的 CDS 来说,本列指定下一个密码子开始的位置。每 3 个核苷酸翻译一个氨基酸,从 0 开始,CDS
的起始位置,除以 3,余数就是这个值,,表示到达下一个密码子需要跳过的碱基个数。该编码区
第一个密码子的位置,取值 0,1,2。0 表示该编码框的第一个密码子第一个碱基位于其 5'末端;1
表示该编码框的第一个密码子的第一个碱基位于该编码区外;2 表示该编码框的第一个密码子的第
一、二个碱基位于该编码区外;如果 Feature 为 CDS 时,必须指明具体值。);
9) attributes:一个包含众多属性的列表,格式为“标签=值”(tag=value),标签与值之间以
空格分开,且每个特征之后都要有分号;(包括最后一个特征),其内容必须包括 gene_id 和
transcript_id。以多个键值对组成的注释信息描述,键与值之间用“=”,不同的键值用“;GTF 文件可以通过从 ensembl 网站或者 ucsc 网站下载:
#人基因组 gtf 文件
wget
http://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.
gtf.gz或者通过 gffread 软件,将 gff 转换为 gtf,该软件来自于 cufflinks 软件包中。
#gff2gtf
gffread my.gff3 -T -o my.gtf
#gtf2gff
gffread merged.gtf -o- > merged.gff3写在最后:有时间我们会努力更新的。大家互动交流可以前去论坛,地址在下面,复制去浏览器即可访问,弥补下公众号没有留言功能的缺憾。原地址暂未启用(bioinfoer.com)。
sx.voiceclouds.cn有些板块也可以预设为大家日常趣事的分享等,欢迎大家来提建议。