我布置了一个作业,让大家可以尝试把cox可以火山图为什么gsea结果不行 这个里面的数据集 GSE101668 ,里面的表达矩阵,进行热图可视化,很多同学完成了作业,我随机挑选其中一个学徒的优秀笔记跟大家分享!
学徒笔记分享
接到曾老师作业后,我兴高采烈的打开GEO网站搜索GSE代号,粗略看一下分组后,打开R语言,想直接用代码下载数据。一顿操作.

竟然无法读取数据??

再仔细一看网站发现,这个原来是一个illumnia NGS的文件
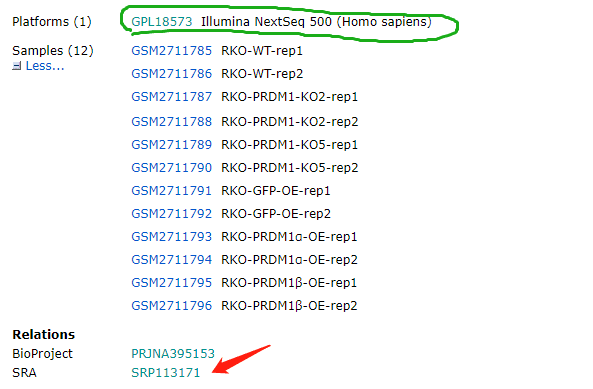
这种文件对于我这个才学了1个多星期的超级菜鸟来说,简直是晴空霹雳。遮盖咋整?
不要慌,根据上次的经验,我们还可以通过原始数据来获得表达矩阵,先去生信树公众号里面搜。然后我看到了曾老师的帖子。

然后我尝试着找文件
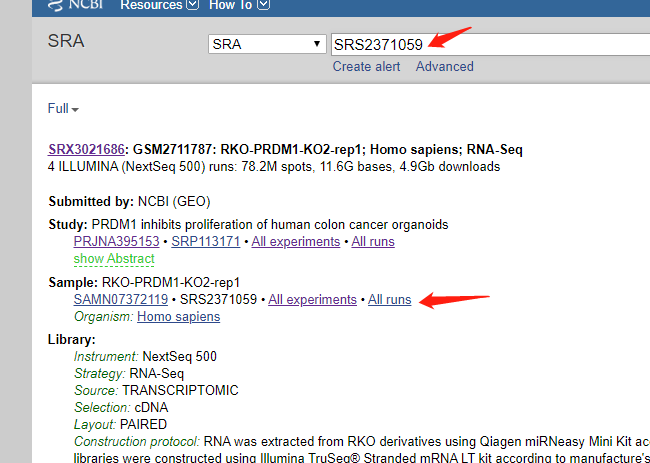
一共12个文件,然后去根据SRR号码去 FTP site里面找
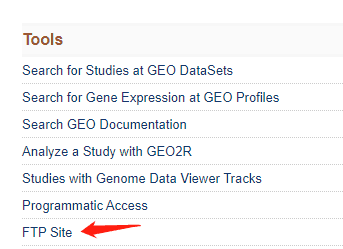
竟然没有数据
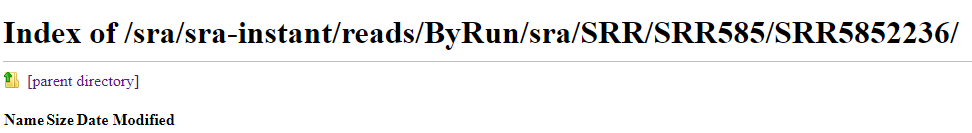
这个就让我懵逼了,为什么没有数据?其实饶了一大圈,我们可以直接从GEO网页直接下载RAWdata
不过这个过程有不错,让我学习了很多以后Linux可能会用到的知识。
我们下载下来后解压缩,发现里面有2组数据,一组是count.txt文件,还有一组是fpkm文件
先试试能不能读取fpkm,因为这个是经过标准化后的数据
library(rio) library(data.table) library(readr)
x1<- fread("GSM2711785_WT1.genes.fpkm_tracking.gz") #其实import是无法读取的
class(x1)
竟然是有2个类型(PS: 因为对fread函数不熟悉,不知道可以加上一个参数)
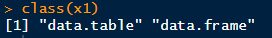
有fpkm还有geneid,有点小激动!
可是仔细一看,他并没有不同组别的数据,这是一个总的或者已经处理的数据,所以对于我来说不需要。
然后我就手动解压缩了counts文件,因为我还是菜鸟,我不太会高级的语言, 我都是一步一步做出来,然后有意思的就是每个分组的数据基因排序都是一样的,所有我们就可以直接cbind
1.获取表达矩阵
下面的代码没有写循环,我愧对老师的教导!
library(rio) library(data.table) library(readr) Wt1<- import("GSM2711785_WT1.counts.genes.txt") Wt2 <- import("GSM2711786_WT2.counts.genes.txt") wt<- cbind(Wt1,Wt2) rownames(wt) <- wt[,1] wt<- wt[,-c(1,3)] colnames(wt) <- c("GSM2711785","GSM2711786")Ko21<- import("GSM2711787_KO2-1.counts.genes.txt")
Ko22 <- import("GSM2711788_KO2-2.counts.genes.txt")
ko<- cbind(Ko21,Ko22)
rownames(ko) <- ko[,1]
ko<- ko[,-c(1,3)]
colnames(ko) <- c("GSM2711787","GSM2711788")ko51 <- import("GSM2711789_KO5-1.counts.genes.txt")
ko52 <- import("GSM2711790_KO5-2.counts.genes.txt")
ko5 <- cbind(ko51,ko52)
rownames(ko5) <- ko5[,1]
ko5 <- ko5[,-c(1,3)]
colnames(ko5) <- c("GSM2711789","GSM2711790")G1 <- import("GSM2711791_GFP1.counts.genes.txt")
G2 <- import("GSM2711792_GFP2.counts.genes.txt")
g <- cbind(G1,G2)
rownames(g) <- g[,1]
g <- g[,-c(1,3)]
colnames(g) <- c("GSM2711791","GSM2711792")p11 <- import("GSM2711793_PRDM1a-1.counts.genes.txt")
p12 <- import("GSM2711794_PRDM1a-2.counts.genes.txt")
p1 <- cbind(p11,p12)
rownames(p1) <- p1[,1]
p1 <- p1[,-c(1,3)]
colnames(p1) <- c("GSM2711793","GSM2711794")p21 <- import("GSM2711795_PRDM1b-1.counts.genes.txt")
p22 <- import("GSM2711796_PRDM1b-2.counts.genes.txt")
p2 <- cbind(p21,p22)
rownames(p2) <- p2[,1]
p2 <- p2[,-c(1,3)]
colnames(p2) <- c("GSM2711795","GSM2711796")dat <- cbind(wt,ko,ko5,g,p1,p2) #这里 将所有数据 组合
rdat <- as.matrix(dat)
rdat[1:5,1:5]
write(rdat,"rdata.csv")
class(rdat)
dim(rdat)
boxplot(rdat)
这个boxplot图不行,我们对他进行取log
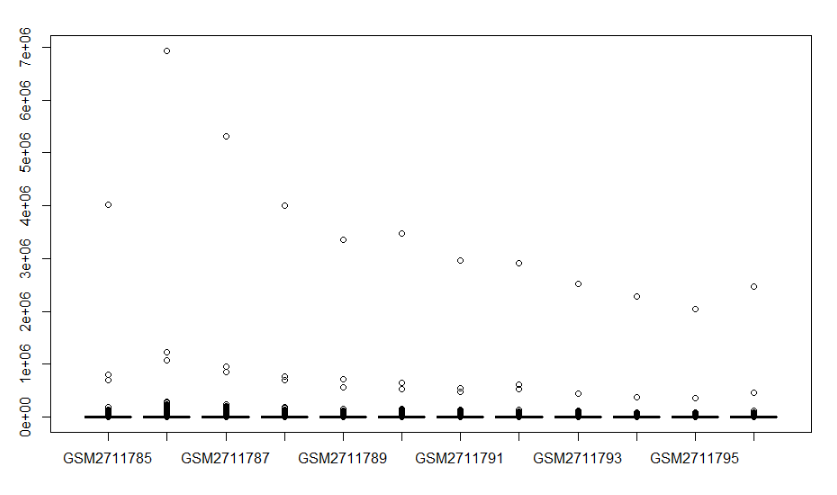
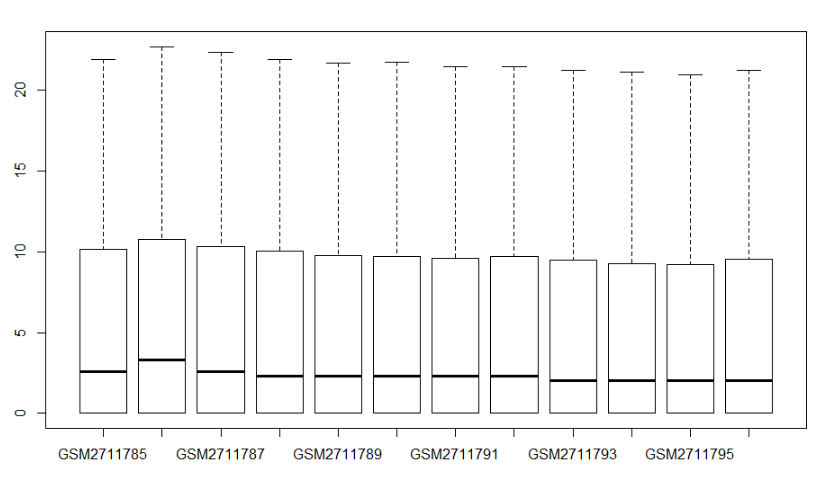
rdathp <- log2(rdat+1)
boxplot(rdathp)
是不是好多了?
由于没有临床信息的文件,那我们就自己创建一个
group_list <- c("WT","KO","OE")
pd1 <- data.frame(GSM=c("GSM2711785","GSM2711786","GSM2711787","GSM2711788","GSM2711789","GSM2711790","GSM2711791","GSM2711792","GSM2711793","GSM2711794","GSM2711795","GSM2711796"),
group= c("WT","WT",rep("KO",4),rep("OE",6)),
subgroup=c("WT1","WT","KO2","KO2","KO5","KO5","GFPOE","GFPOE","PRDM1a","PRDM1a","PRDM1b","PRDM1b"))
save(rdat,rdathp,pd1,file="h1.Rdata")
我居然不解释为什么呀log,我错了!而且我分组里面,居然使用了这么丑陋的代码
2.group_list(实验分组)
rm(list = ls()) load(file = "h1.Rdata") library(stringr)
#分组
pd1[1:12,1:3] #查看pd 找到组别
不愧是自己创建的,真整齐划一
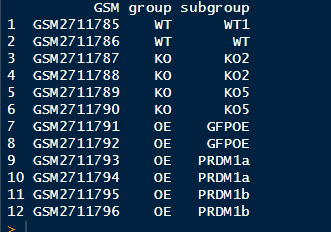
library(stringr) #设置group
group_list=ifelse( str_detect(pd1group,"WT"),"WT",ifelse(str_detect(pd1group,"KO"),"KO","OE"))
group_list = factor(group_list,
levels = c("WT","KO","OE"))
#忘记给pd1 rownames
rownames(pd1) <- pd1[,1]
p = identical(rownames(pd1),colnames(rdathp));p
当然输出是TRUE
dim(rdathp)
dim(pd1)
save(group_list,rdat,rdathp,pd1, file = "h2.Rdata")
3.画热图
rm(list = ls())
load(file = "h2.Rdata")library(RColorBrewer)
#热图
cg=names(tail(sort(apply(rdathp,1,sd)),500))#apply按行('1'是按行取,'2'是按列取)取每一行的方差,从小到大排序,取最大的500行
n=rdathp[cg,] #形成一个新的矩阵,只有那排名500的基因#绘制热图
annotation_col=data.frame(group=group_list)
rownames(annotation_col)=colnames(n) #这里两步就是设置一个分组列表
library(pheatmap)
pheatmap(n,
show_colnames =F,
show_rownames = F,
annotation_col=annotation_col,
scale = "row" )
dev.off()
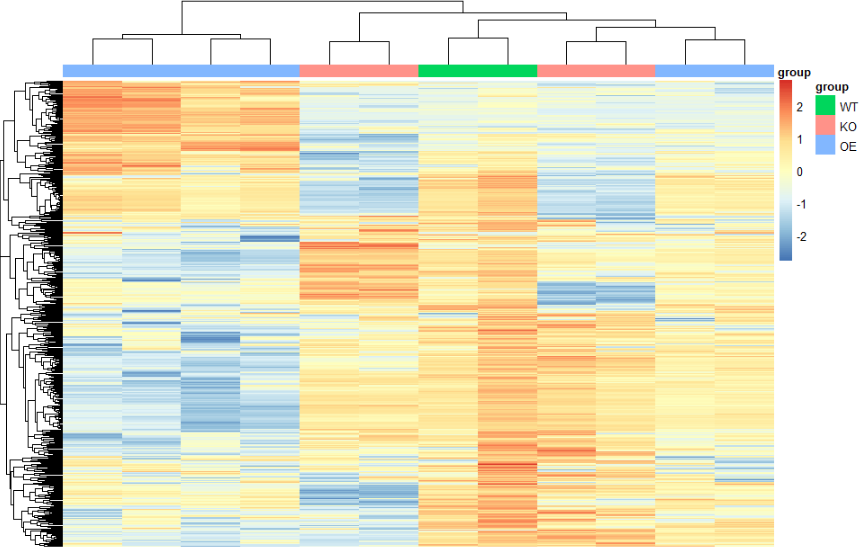
- 进行颜色变换
color.1 <- colorRampPalette(rev(brewer.pal(n = 7, name ="RdYlBu")))(100)
color.3 <- colorRampPalette(rev(c("#ff0000", "#ffffff", "#0000ff")))(500)
color.2 <- colorRampPalette(rev(c("#ff0000", "#000000", "#00ff00")))(500)
pheatmap(n,
show_colnames =F,
show_rownames = F,
annotation_col=annotation_col,
scale = "row",
color = color.1) #颜色这里可以自行调整
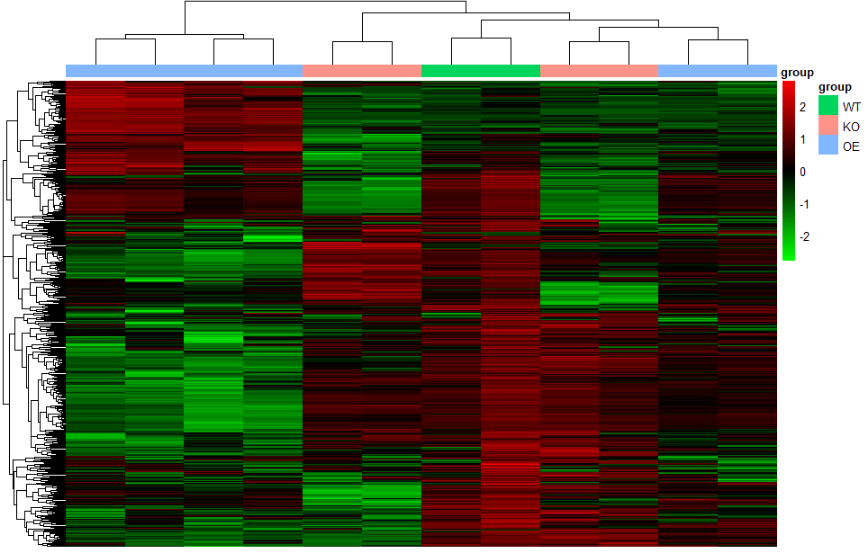
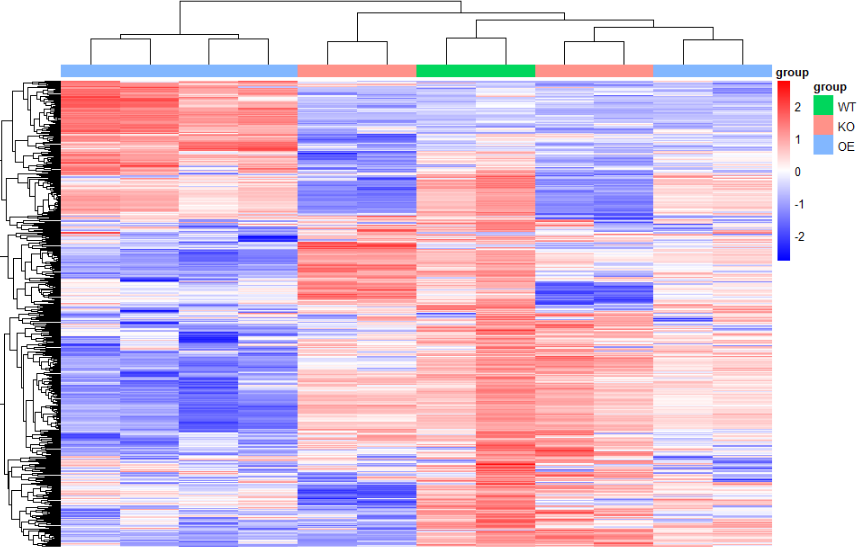
- 切割,同时可以对treeheight 进行修改
p <- pheatmap(n,scale = "row",treeheight_row = 5 ,cutree_rows=5,color = color.2)
class(p)
cluster_infor <- data.frame(labels=ptree_collabels,order=cutree(p$tree_col,2))
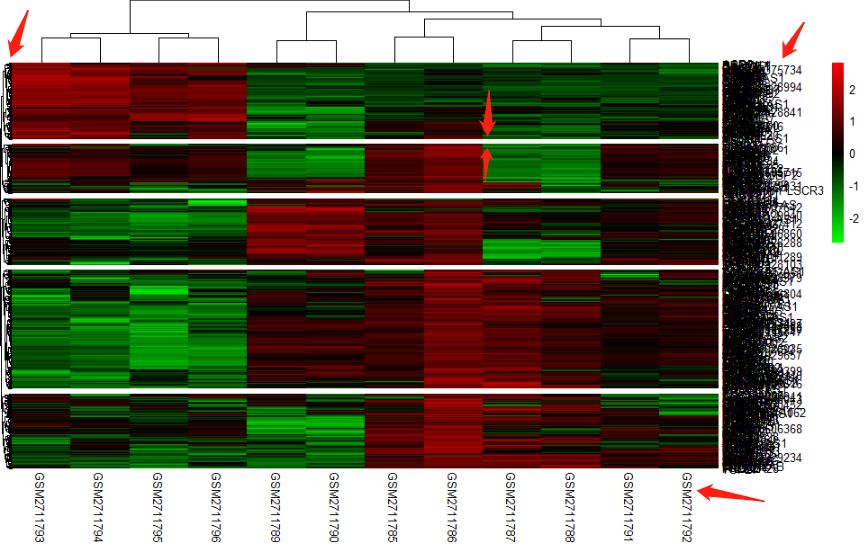
还可以输出分组,缺图, 算了吧
- 最后是输出图像
pdf(file = "heatmap.pdf",height = 8,width = 6)
print(p)
dev.off()其实也可以ggsave进行输出